
Wan YBA1,20, Simpson MA2,20, Dulfer E23, Osborn DPS1, Kerstjens M23, Vos Y23, Pinard A5, Regalado E5, Guo DC5, Boileau C6,7,8,9, Jondeau G6,7,8,10, Benarroch L6, Loeys B11, Luyckx I11, Van Laer L11, Verstraeten A11, DeBacker J12, Ramachandran V1, Ashraf QM3, Lau L3, Garcia M1, Gaer J24, Bharj J1, Sneddon J13, Fisher E14, Dean J15, Isekame Y1, Saggar A16, Milewicz D5, Jahangiri M17,18, Behr ER18, Child A1,4,Smith A19,21, Aragon-Martin JA1,3,21.
1) Genetics Research Centre, Molecular and Clinical Sciences Research Institute, St. George’s University of London, United Kingdom; 2) Division of Genetics and Molecular Medicine, Kings College London, United Kingdom; 3) NHLI, Imperial College, , London, United Kingdom; 4) Marfan Trust, Guy Scadding Building, London, United Kingdom; 5) Department of Internal Medicine, Division of Medical Genetics, University of Texas Health Science Center at Houston McGovern Medical School, Texas, USA; 6) Laboratory of Vascular Translational Science, INSERM U1368, Paris, France; 7) Centre de référence pour le syndrome de Marfan et apparentés, APHP hospital Bichat, Paris, France; 8) Université Paris Diderot, Paris France; 9) Department de Genetique, APHP, Hopital Bichat Paris, France; 10) Service de Cardiologie, APHP, Hopital Bichat, Paris, France; 11) Centre of Medical Genetics, Faculty of Medicine and Health Sciences, University of Antwerp, Belgium; 12) Adult Congenital Heart Disease and Cardiogenetics, Ghent University Hospital, Ghent, Belgium; 13) East Surrey Hospital, Redhill, Surrey, United Kingdom; 14) The Park Surgery, Albion Way, Horsham, Surrey, United Kingdom; 15) Clinical Genetics, NHS Grampian, Foresterhill, Aberdeen, United Kingdom; 16) Clinical Genetics Unit, St. George’s University Hospitals NHS Foundation Trust, United Kingdom; 17) Department of Cardiothoracic Surgery, St. George’s University Hospitals NHS Foundation Trust, United Kingdom; 18) Cardiology Academic Group, Cardiology Department, St. George’s, University Hospitals, NHS Foundation Trust, United Kingdom; 19) Academic Department of Vascular Surgery, Cardiovascular Division, BHF Centre of Research Excellence, Kings College London, United Kingdom; 20) These authors contributed equally to this work; 21) These authors contributed equally to this work. 23) Clinical Genetics, University Medical Center Groningen, The Netherlands. 24) Royal Brompton & Harefield Hospitals NHS Foundation Trust, London, United Kingdom.
Key words: FTAAD; LMOD1; smooth muscle contraction; WES; variants; Familial Thoracic Aortic Aneurysm and Dissection; diagnosis.
Subject codes: Basic Translational, and Clinical Research- Animal Models of Human Disease and Basic Science Research ; Epidemiology, Lifestyle, and Prevention-cardiovascular disease; Genetics- Genetically Altered and Transgenic Models;
Vascular disease- Aneurysm, Aortic Dissection.
Corresponding author’s contact information
Dr Anne Child MD FRCP, Marfan Trust, B119 – Guy Scadding Building
Imperial College, Dovehouse Street, London SW3 6LY, United Kingdom
annechildgenetics@outlook.com; Tel: +44 (0)7554694400
Received 6 August 2020; revised 25 February 2022; accepted 26 May 2022
Abstract
Objective
Thoracic Aortic Aneurysm & Dissection (TAAD) can arise at any time without previous warnings and with fatal consequences. We seek to determine the genetic cause of TAAD in a UK multi-generational Caucasian family.
Approach
Whole exome sequencing was performed and analysed on three affected individuals. Both in vitro and in vivo studies evaluated functionally the role of the candidate gene.
Results
Twenty-five variants in LMOD1 were identified from 1751 probands from UK and international cohorts, of which six were identified to be most deleterious by in silico validation. N-terminus variants in the TMBS domain & C-terminus variants in the WH2 domain were enriched in their respective domains in the complete TAAD patient cohort versus the gnomAD genetic database. Myofibroblasts from the proband with variant Val595Ala demonstrated reduced nucleation of actin filaments, mislocalization of LMOD1 protein, and impaired contractility. Knockdown of paralogs lmod1a/lmod1b in zebrafish demonstrated delayed development of the aortic precursors (pharyngeal arches), rescued by co-injections with wild-type LMOD1 mRNA. Conversely, this zebrafish knockdown could not be fully rescued by the mutant c.1784T>C [p.(Val595Ala)] containing LMOD1 mRNA, strongly suggesting this variant is pathogenic for TAAD.
Conclusions
This study suggests that variant Val595Ala predisposes to TAAD due to abnormal LMOD1 functionality. Future variants identified in the WH2 domain may also delay actin polymerization, compromising actin length, dynamics and interaction with myosin in the vascular smooth muscle contraction pathway. This study strongly supports a role for LMOD1 in development and maintenance of smooth muscle through its control of the cells’ actin cytoskeleton.
Introduction
Thoracic Aortic Aneurysm and Dissection (TAAD) is a severe disease with an increasing incidence, currently at 9-15 per 100,000 patient-years16 and accounting for approximately 15,000 deaths per year in the USA17. TAAD is typically inherited in an autosomal dominant manner and even within affected kindreds the disorder shows variability in penetrance and severity, with notably increased severity in males18-20.
TAAD can be present in syndromic or non-syndromic form. The most common and best characterized syndromic TAAD is Marfan syndrome, which also demonstrates a number of other systemic features such as ectopia lentis and is clinically diagnosed using the revised Ghent criteria21 including identification of an FBN1 variant.
In non-syndromic forms of TAAD, a quarter of cases are familial (FTAAD)22, strongly suggesting a genetic basis in the presence of a normal clinical phenotype. The greatest contributor to non-syndromic TAAD, in approximately 14% of families is the gene ACTA223. Variants in ACTA2 are also reported to be linked to other smooth muscle cell disorders like syndromic TAAD, Coronary Artery Disease (CAD), ischemic stroke, and Moyamoya disease (MMD)24. Biallelic variants in MYH115,6,8, MYLK1 and LMOD112 are reported to cause Megacystis Microcolon Intestinal Hypoperistalsis Syndrome (MMIHS), a congenital disorder characterized by smooth muscle disfunction affecting the bowel and bladder; MYH117,9-11 and MYLK2-4,15 are also known as TAAD-causing genes because the heterozygote variants in these two genes are associated with TAAD. The underlying molecular genetics of TAAD is heterogeneous, with many genes reported to date as sites of pathogenic variants in syndromic and non-syndromic TAAD2-4,7,9,10,23,25-50. Investigations into the biological roles of this group of genes have revealed common pathways. Variants in the genes involved in signalling of TGF-β and in contraction of smooth muscle cells develop TAAD24,51-53, which are the two major pathways involved in TAAD. However, many families do not have a causative variant in the reported TAAD genes (as much as 84.6% [see figure S4.1 in Supplement 4]), suggesting that additional FTAAD genes are yet to be found. To extend the understanding of the genetic basis of TAAD, a whole exome sequencing (WES) approach was used in a large multigenerational family of British ancestry (M441) with autosomal dominant inheritance of TAAD.
Material and methods
Subjects
All individuals in the UK study were ascertained through the Aortopathy clinic at St George’s University Hospitals Foundation NHS Trust. Ethical consent was obtained from the National Research Ethics Service Committee London- Bloomsbury (reference; 14/LO/1770, project ID; 99752). A license was granted for the storage of samples under section 16 (2) (e) (ii) of the Human Tissue Act 2004 (No. 12335). Patient and family members have read and signed the relevant consent forms in this study.
Sequencing and variant interpretation tools
WES was used to identify the causative variants in the study family (M441) by generating exome variant profiles for the proband (IV:5) and two distantly affected relatives (IV:2 and IV:16) (figure 1A). It was performed at the KCL-GSTT BRC Genomics facility at Guys Hospital, London as a collaborative research service. Exome libraries were generated using ‘Agilent SureSelect All exon 50Mb capture’ on an Agilent Bravo Liquid handling robot and exome enriched libraries were sequenced by paired-end reads on an Illumina HiSeq2000 platform and interpreted using an in-house pipeline54. Subsequent variants detected were further filtered as previously reported55. Sanger sequencing was performed to validate variants identified in affected family members.
Variants were assessed using in silico prediction tools and recommended tools56. The Genome Aggregation Database (gnomAD v3.1.2)57 was used to determine the frequency of variants identified in a large population database. Two recurrent variants previously reported to be pathogenic [c.492C>T p.(Arg149Cys) and c.819C>T p.(Arg258Cys)]24 in gene ACTA2 (NM_001613.2) for FTAAD were found to be present on gnomAD at a frequency of 0.000007. As causative variants in the ACTA2 gene were the main contributor to TAAD, therefore variants identified in our cohort with a higher frequency than the most common ACTA2 causative variants were further filtered out as they would be expected to be rarer. Variants identified in this study were assessed in known domains of their protein, tissue specific gene expression, and localization within the cell58-61.
In vitro and in vivo studies
Patient and age- and sex- matched control skin biopsies were cultured and differentiated to myofibroblasts by a 72h treatment with TGFβ113. For immunofluorescence studies, cells were processed with α-SMA (mouse monoclonal, A5228, Sigma-Aldrich) and LMOD1 (rabbit polyclonal, 15117-1-AP, Proteintech) as primary antibodies and followed by secondary goat anti-Mouse IgG Alexa Fluor 488 (A-11001, Invitrogen) for α-SMA and goat anti-Rabbit IgG Alexa Fluor 488 (A-11008, Invitrogen) for LMOD1 and co-stained with Texas Red™-X Phalloidin (T7471, Invitrogen) and DAPI were obtained from a confocal inverted microscope DMi8 from Leica. Protein quantity was assessed by Western-blot analysis using the same primary antibodies and a standard protocol13,14. Collagen gel assays were used to assess the contractility of patient versus control myofibroblasts13,14 (Supplement 2).
Wild-type (AB x Tup LF) and Tg (kdrl:EGFP) s843Tg (Zebrafish International Resource Centre) zebrafish were maintained and staged as previously described by Westerfield et al62. All fish lines are maintained by the Biological Research Facility at St George’s University of London in a recirculating system (Tecniplast) at 28.5°C, on a 14/10-hour light/dark cycle and fed with brine shrimp three times per day. Knockdown of lmod1a/lmod1b was achieved by use of antisense morpholino oligonucleotides targeting the splice site (Supplement 1). An optimized dose of 4ng/ul of lmod1a and lmod1b was co-injected under the blastomere of 1 to 2-cell embryos using a pulled borosilicate glass needle with a standard microinjection system.
Zebrafish and human wild-type mRNAs were used to study overexpression and rescue of the MO-induced phenotype. Full-length wild-type LMOD1 cDNA was synthesized (Genscript, USA) and cloned into a PCS2+ vector at restriction sites BamH1 and Xho1. Site directed mutagenesis (Agilent) was utilized to recreate variants identified in the probands and families in this study, using manufacturers recommended protocols. Wild-type and mutant LMOD1 containing vectors were transformed (One Shot® TOP10 chemically competent E. coli Life technologies), colonies were grown & cultured in ampicillin-containing LB agar/broth, isolated (Miniprep kit, Qiagen) and Capped RNA was synthesized (Ambion) using manufacturer’s recommended protocol. RNA was normalized to 100ng/ul and injected as 50pg into the cytoplasm of 1-cell staged embryos prior to injection of MOs.
Aortic arch measurements
Wild-type and mutant embryos (AB x Tup LF and Tg(kdrl:EGFP)) were fixed in 4% paraformaldehyde for 20 minutes and mounted with glycerol onto slides for light microscopy or 1% low melt agarose onto 35mm cell culture glass bottom dishes for confocal microscopy. Images were taken using a stereomicroscope SMZ 1000 (Nikon), fluorescence stereomicroscope MZ16F (Leica) and confocal microscope LSM510 Meta (Zeiss). Images were processed with Adobe Photoshop CC (Adobe Systems) using the same settings to maintain a genuine contrast of samples. ImageJ was used to measure the aortic arches in transgenic (kdrl:EGFP) zebrafish by outlining each of the arches and using the software measurement tool to calculate the
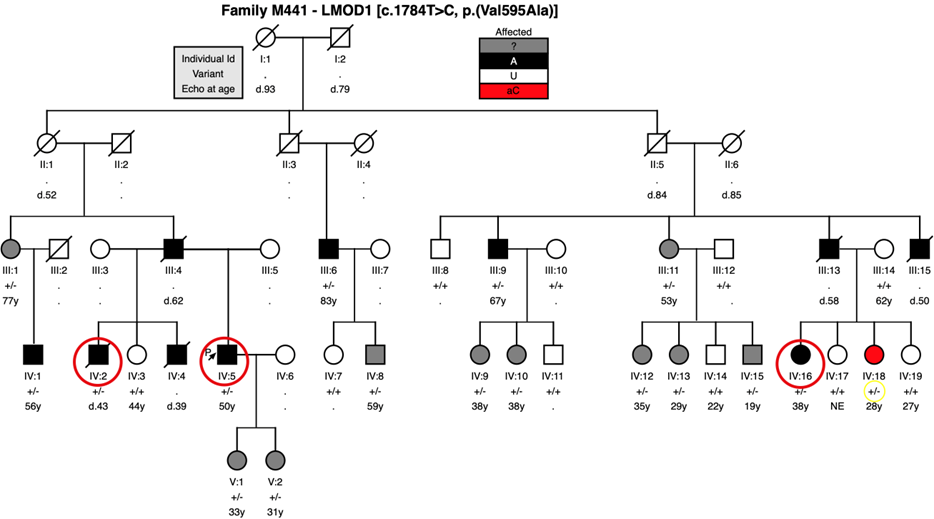
Figure 1A- Family M441 pedigree where the LMOD1 variant c.1784T>C [p.(Val595Ala)] was identified to segregate with disease, and incomplete penetrance in apparently unaffected patient IV:18 (shaded in red and genotype circled in yellow) that was classified as asymptomatic carrier. Affected patients IV:2, IV:5 and IV:16 (circled in red) were studied in 2014 in a WES analysis. Patients III:1, III:11, IV:8, IV:9, IV:10, IV:12, IV:13, IV:15, V:1 and V:2 (shaded in grey) were classified as ambiguous phenotype = patients who did not had detectable aortic dilation by transthoracic echo but they presented with two or more minor features associated with a marfanoid phenotype. Notes: . = No Data (individuals who were not Sanger sequenced); . = Not Evaluated (deceased patients will be prefixed “d.” with the age of death
surface area. Statistical analysis (GraphPad Prism) by Tukey’s multiple comparisons test was undertaken to test for any significant differences between the measurements of each study group.
Results
The proband IV:5 in family M441 (figure 1A) initially consulted a cardiologist as both his father (III:4) and half-brother (IV:2) died as the result of type A dissection involving the ascending aorta. Doppler transthoracic echocardiography measurements revealed he had a mildly dilated aortic
root at 4.2cm (Z-score = 2.28)63,64. Clinical investigation of other family members revealed five living relatives (III:6, III:9, IV:1, IV:5, and IV:16) who had previously undergone surgery for TAAD or who had a dilated aortic root. Autopsy report revealed aortic dissection as the cause of death for four males (III:4, III:13, III:15, and IV:4) (mean age of death 52.25 years, range 39-62 years).
Due to a lack of a genetic diagnosis in this family, WES was undertaken for patients IV:2, IV:5 and IV:16. Initial analysis of exome variant call format (VCF) files examined changes in 53 known TAAD genes65,66, including the recent THSD4 gene50. No pathogenic variants were identified in any of the three affected individuals and therefore known genetic causes were excluded.
Further analysis revealed five candidate variants, which Sanger sequencing validation rule out two as artifacts, and the other three were rare (MAF ≤0.00003), protein-altering, heterozygous variants present in the profiles of each of the three sequenced affected patients in the study family (M441). The three candidate variants located in LMOD1, SPHKAP and ZNF483 were each evaluated for co-segregation with the trait in the pedigree by Sanger sequencing. Neither the variant in SPHKAP nor the variant in ZNF483 co-segregated with the disease.
LMOD1 variant c.1784T>C, p.(Val595Ala) segregates with disease in a large British family
The c.1784T>C, p.(Val595Ala) (ENST00000367288/NM_012134) allele in LMOD1 was carried by each of the five affected living patients (figure 1A) and was also present in the DNA sample obtained from a deceased affected half-brother of the proband (IV:2).
A further 10 heterozygous individuals were identified in the pedigree who had an ambiguous phenotype, meaning that none of them had detectable aortic dilation by transthoracic echo but they presented with two or more minor features associated with a marfanoid phenotype21. One heterozygous clinically unaffected individual was found who did not present with a detectable aneurysm. However, this individual (IV:18, aged 28 at the time of examination) was documented as having an isolated high arched palate, a feature shared with the proband and 10 other heterozygous individuals.
A parametric linkage analysis of the observed segregation of the LMOD1 c.1784T>C allele and the affection status, generated a LOD score in excess of 2.3, p=0.0005 (LOD score 6.7 was observed from an affected-only analysis, dominant model, disease frequency 0.0001, and with penetrance 0.9). These data suggest that the allele located in LMOD1 was likely to be causative in this family and should therefore be investigated further.
The c.1784T>C LMOD1 variant is located in the third and terminal exon of the encoded 3927bp transcript, and the predicted p.(Val595Ala) substitution in the mature protein product replaces a conserved valine residue. This residue is located in the C-terminus WH2 domain known to be an actin-binding domain (figure 2). The c.1784T>C allele was not observed in gnomAD and was not seen in ~5,500 exomes (in-house samples) sequenced and processed using the same laboratory protocols and data processing pipeline as used in the current study54. The rarity of this allele suggests that it is likely to have a role in the pathogenesis of aortic aneurysm in this population of TAAD patients.
Copy Number Variation (CNV) was assessed in the UK TAAD family M44ue1 by exome depth software and rare and previously unreported deletions, or duplications were considered candidates. There was no evidence of co-segregation in the 3 individuals (IV:2, IV:5 and IV:16) (figure 1A) of any structural variation with the phenotype.
LMOD1 variants in UK and international TAAD cohorts
The possibility that further LMOD1 alleles may explain additional cases of TAAD was explored by Sanger sequencing of the three LMOD1 exons and their associated splice sites in a UK cohort of 91 unrelated individuals with TAAD, as well as screening for LMOD1 variants in exome profiles of a total of 1660 international sporadic and familial TAAD probands; from Houston, Texas, USA (780 probands à 448 FTAAD families and 332 TAAD sporadic), Paris, France (45 FTAAD families), Antwerp, Belgium (790 FTAAD families), Ghent, Belgium (44 FTAAD families), and Netherlands (1 FTAAD family – figure 1B), which revealed further variants in this gene (Table 1A). All 780 probands from the USA cohort were screened by WES, and rare LMOD1 variants (MAF ≤0.005 in the ExAc database) were shared with this study. Altogether, twenty-five variants were detected across all three exons of LMOD1, eight of which were found to be rare (frequency ≤0.00001), high conservation score (PhastCons 100 ≥0.9 & Phylop 100 ≥2), scoring in the top 1% of most deleterious substitutions (CADD ≥20), and classified as damaging in the majority of in silico tools used (Table 1C). Rare (frequency ≤0.00001) WH2 domain variants were enriched in the complete TAAD patient cohort compared with that in gnomAD (p<3.077e-15) (see supplement 5 – part 3).
LMOD1 variant c.25C>T, p.(Arg9Trp) found in the Belgium-Antwerp TAAD cohort
It was identified in a proband originally from Lübeck, Germany, and classified with bicuspid aortic valve & thoracic aortic aneurysm phenotype. No family members were available for family segregation.
LMOD1 variant c.64G>T, p.(Glu22Ter) found in the American TAAD cohort
It was identified in the proband (Type A dissection) and her mother after a Trio-Study. We do not have clinical records on the mother at this moment (age 64 in 2020), however, it is interesting that the maternal grandfather had an aortic aneurysm.
LMOD1 variant c.164C>T, p.(Thr55Met) found in the Belgium-Antwerp TAAD cohort
It was identified in a male proband born in 1957 and classified with aortic aneurysm phenotype (4.5cm). The family history includes a causative variant in gene TTN in uncle (frameshift mutation – National Centre for Crisis Management – NCCM), and the proband’s father died of Acute Myocardial Infarction (AMI) at age 67. Proband was TTN negative. No family members were available for family segregation.
LMOD1 variant c.1723C>T, p.(Arg575Cys) found in the American TAAD cohort
It was identified in a proband classified with Early-Onset Sporadic Thoracic Aortic Dissection (ESTAD). No family members were available for family segregation.
LMOD1 variant c.1769T>C, p.(Leu590Pro) segregates with disease in a large Dutch family
Affected patient III:2 from the Dutch TAAD family Netherlands01 was studied in 2015 in a Dutch TAAD-panel version 2 (figure 1B). Affected patient III:2, his unaffected wife and affected son IV:2 were studied in 2020 in a WES trio-analysis which resulted in this LMOD1 variant. No CNV analysis was done in the WES study. The c.1769T>C, p.(Leu590Pro) allele in LMOD1 was carried by each of the five affected patients (figure 1B). Affected patients dissected at least once; IV:2 had a Type A dissection in 2019 after screening performed (Echo/MRI) in 2015 with normal diameters. This variant was also present in three apparently unaffected patients (III:6, IV:3, and IV:5) that were classified as asymptomatic carriers. All three asymptomatic carriers had no aortic dilatation/dissection observed when screened by Echo/CT-scan: III:6 (2015 & 2021); IV:3 (2015 & 2020); and IV:5 (2014, 2017 & 2020).
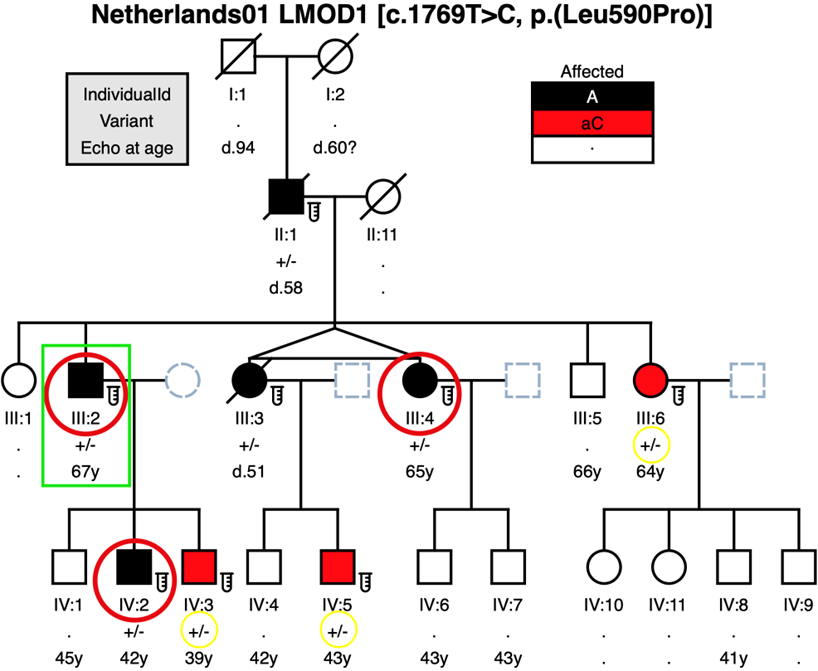
Figure 1B- Family Netherlands01 pedigree where the LMOD1 variant c.1769T>C [p.(Leu590Pro)] was identified to segregate with disease, and incomplete penetrance in patients III:6, IV:3 and IV:5 (shaded in red and genotype circled in yellow) that were classified as asymptomatic carriers. Affected patient III:2 (square in green) was studied in 2015 in a TAAD-panel version 2 (ACTA2, COL3A1, EFEMP2, ELN, FBN1, FBN2, MYH11, MYLK, NOTCH1, PLOD1, PRKG1, SCARF2, SKI, SLC2A10, SMAD2, SMAD3, SMAD4, TGFB2, TGFB3, TGFBR1, TGFBR2). Affected patient III:2, his unaffected wife and affected son IV:2 were studied in 2020 in a WES trio-analysis. Notes: . = No Data (individuals who were not Sanger sequenced); . = Not Evaluated (deceased patients will be prefixed “d.” with the age of death).
WH2 domain variants
The variant Arg575Cys changes the residue at that position from positive to negative charge, the new residue is more hydrophobic & smaller than the wild-type and might introduce a new disulfide bond, all these points can result in loss of bonds and/or disturb the correct folding of the protein, which can cause loss of interactions with other molecules or residues; the variant Leu590Pro changes to a smaller residue and introduces a completely new amino acid structure; and the variant Val595Ala changes slightly to a smaller residue.
The wild-type of these 3 variants occur often at their position in the protein sequence. Other residues have also been observed, but these 3 mutant residues were not observed among the other residue types observed at their position in other, homologous sequences. All 3 variants are located near a highly conserved position, they are in the WH2 domain that is important for binding of other molecules. All these three variants could disrupt the original
protein structure and altered its function, which can lead to changes of interactions with other molecules67.
Mutant LMOD1 cells display mislocalization and reduced contractility.
To determine the in vitro functional and cellular relevance of LMOD1, dermal fibroblast cells from the proband carrying the LMOD1 c.1784T>C variant, and an age- and sex-matched control were differentiated to myofibroblast cells. More cases and control samples were not available for this study. As LMOD1 was previously reported to nucleate actin monomers in smooth muscle restricted cells68, antibodies were utilized to assess changes in the cytoskeleton. A reduction of α-SMA and F-actin (phalloidin) staining (figure 3A) highlights a reduction of actin filaments in the cytoplasm suggesting that the variant in LMOD1 may be limiting the nucleation of actin filaments. Localization of LMOD1 protein was studied by immunofluorescence. Mutant cells demonstrated fluorescence of LMOD1 protein in the perinuclear region compared with diffuse staining in control cells, suggesting LMOD1 is mislocalized because of the patient variant (figure 3B). LMOD1 protein levels were quantified by Western blot analysis using lysates from dermal fibroblast to myofibroblast cell stages. A steady increase in quantity during differentiation was observed demonstrating that LMOD1 protein is required during myofibroblast function (figure 3C). There was no visual difference in quantity of LMOD1 protein between patient and control cells, suggesting that the variant does not impair the production of LMOD1 protein. The activity of the myofibroblasts was assessed using a collagen gel assay, which revealed patient cells were less able to contract in comparison with controls (p=0.03, figure 3D). Combined, these data indicate that mutant LMOD1-containing vascular smooth muscle cells in the aorta may also demonstrate impaired actin cytoskeleton and reduced contractility.
Knockdown of lmod1a/lmod1b in zebrafish causes a global phenotype.
Zebrafish were utilized as an in vivo animal model to assess if morpholino-mediated knockdown of lmod1a/lmod1b (lmod1a-b) can recapitulate the clinical features associated with TAAD. Zebrafish have two paralogs, lmod1a and lmod1b that have resulted from a genome duplication event common to teleost fish69.
Initially, knockdown of each paralog at 4ng/ul separately was undertaken but embryos did not display a phenotype. Both were simultaneously knockdown by injection at 4ng/ul (2ng/ul each) to recapitulate the human phenotype in zebrafish. Out of the co-injected embryos, 97% displayed a spectrum of global phenotypic changes that were also observed in previously reported TAAD genes evaluated in zebrafish26,28. This suggested that there may be overlapping and redundant functions between lmod1a and lmod1b. The phenotype included moderate to severe eye abnormalities, pericardial oedema and tail curvature, and pooling of blood in the yolk sac. The possible effect of non-specific binding of MO was considered, and a standard control MO was injected under the same conditions as the lmod1a-b MOs. One percent of control MOs embryos displayed an abnormal phenotype, consisting of moderate tail curvature compared with 97% in the lmod1a-b co-injected study group. P53 upregulation has been reported to be an
Figure 2
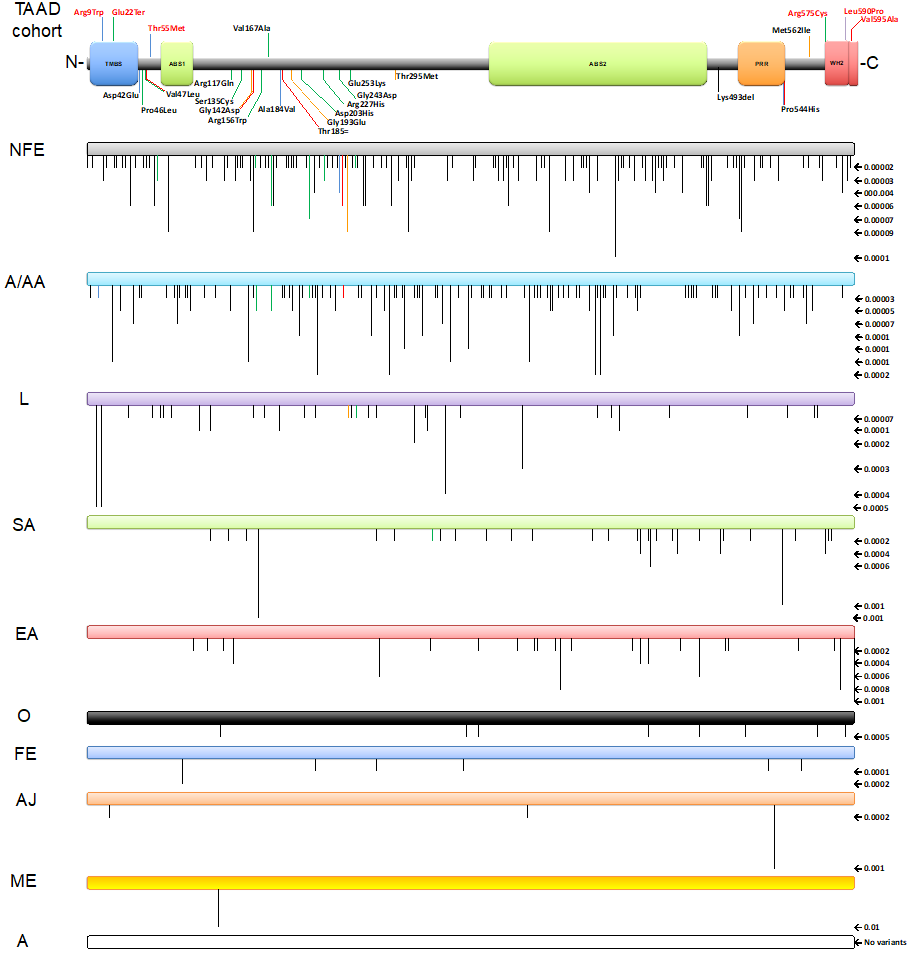
Figure 2. Top bar shows a cartoon representation of the human LMOD1 protein domain architecture showing the 25 variants found in this study (International and UK TAAD cohorts): with upward lines showing changes at the protein level identified to be likely pathogenic, and with downward lines identified to be likely benign. The lines are coloured in black (Belgium-Ghent), blue (Belgium-Antwerp), green (USA), orange (France), purple (Netherlands), and red (UK).
TMBS- Tropomodulin binding site, ABS1- Actin binding site 1, ABS2- Actin binding site 2, PRR- Proline-rich region and WH2- Wiskott-Aldrich homology 2 domain (3rd Actin binding site – ABS3).
Bars below show rare variants (≤0.00006 – from total allele frequency) identified on population database gnomAD (v3.1.2) and separated into different populations with their respective frequencies in numbers shown on the right-hand side and in downward black lines on each bar. When studying individual populations in each variant, amino acid change Thr55Met (0.00003[v3.1.2] – 0.000009[v2.1.1]) & Met562Ile (0.00002[v2.1.1]) were only identified in Non-Finnish Europeans.
A = Amish; A/AA = African/African American; AJ = Ashkenazi Jewish; EA = East Asian; FE = (Finnish) European; L = Latino/Admixed American; ME = Middle Eastern; NFE = (Non-Finnish) European; O = Other; SA = South Asian.
Table 1A

Table 1B
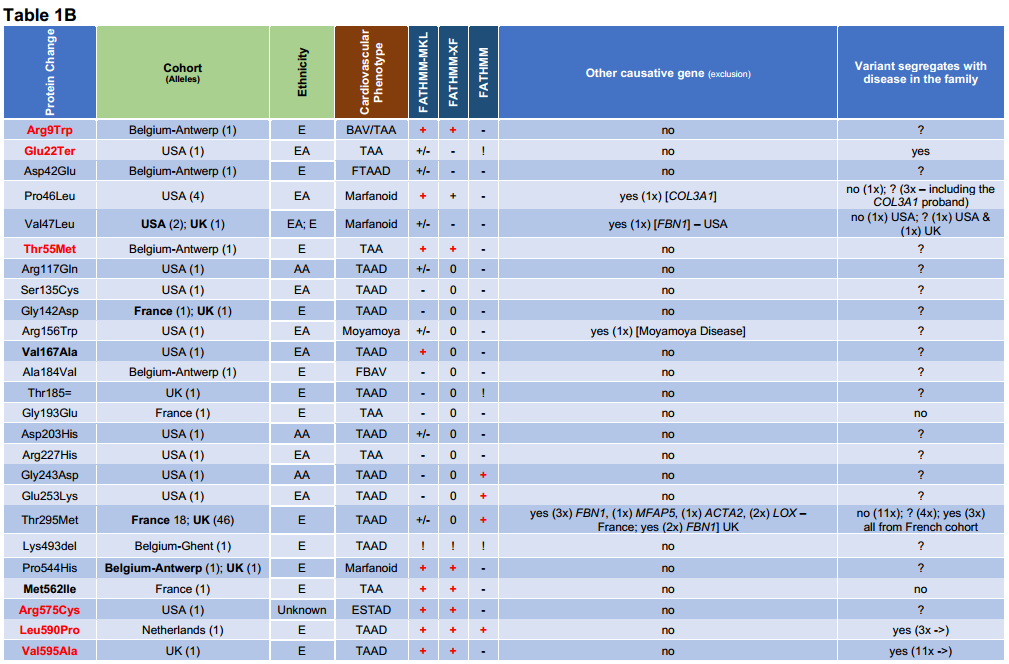
Table 1C
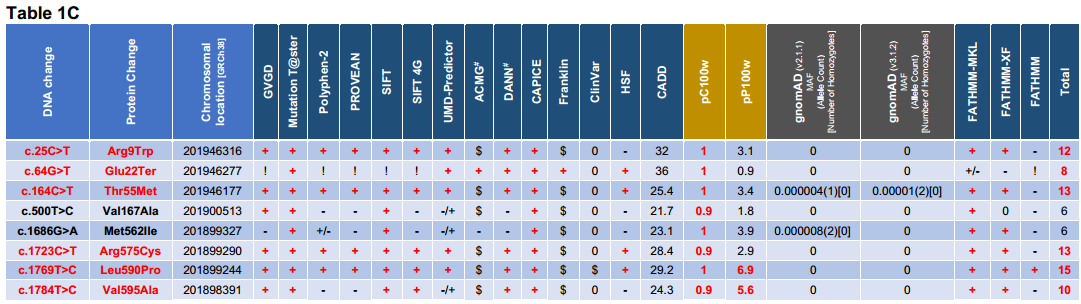
Table1. Variant analysis and interpretation table of LMOD1 (ENST00000367288/NM_012134) alleles detected in 1751 UK and international TAAD probands. Variants coloured in red were identified as likely to be pathogenic based on in silico prediction tools used. 1analysis is from the nucleotide at each location and not from the entire deletion for pC100w & pP100w). #Data taken from Varsome. Scores: (+) = Damaging; (+/-) = Likely Damaging; (-/+) = Likely Benign; (-) = Benign; (!) = n/a because either deletion, synonymous or stop codon; ($) = Uncertain Significance or VUS; (0) = Absent or No prediction found.
Table1A: Databases and in silico tools used [GVGD (Grantham Variation/Grantham Deviation algorithm), MT (Mutation T@ster), PP2 (Polyphen-2), PROVEAN (Protein Variation Effect Analyzer), SIFT (Sorting Intolerant From Tolerant), SIFT4G (Sorting Intolerant From Tolerant 4G), UMD (UMD-Predictor), ACMG (The American College of Medical Genetics and Genomics guidelines for variant interpretation – taken from Varsome), DANN (Deleterious Annotation of genetic variants using Neural Networks), CAPICE (Consequence-Agnostic prediction of Pathogenicity Interpretation of Clinical Exome variations), Franklyn (Franklin by Genoox), ClinVar (public archive of reports of the relationships among human variations and phenotypes from NCBI), Human Splicing Finder (HSF), CADD (Combined Annotation Dependent Depletion score – CAAD GRCh38-v1.6 [Phred-style CADD raw scores are displayed – higher scores are more likely to be deleterious]), pC100w (from UCSC Conservation score file “phastCons100way.UCSC.hg38”), pP100w (from UCSC Conservation score file “hg38.phyloP100way.bw”) and gnomAD (mean allele frequency from v2.1.1 and v3.1.2 was rounded up)]. The rs number was given when available.
Table 1B: Cohort groups showing the number of alleles for each variant. Some variants were found in different cohorts: p.(Val47Leu) USA & UK; p.(Gly142Asp) & p.(Thr295Met) France & UK; and p.(Pro544His) Belgium-Antwerp & UK. Each proband’s ethnicity is indicated when available for each variant (AA = African-American; E = European; EA = European-American). The cardiovascular phenotype is shown: BAV (Bicuspid Aortic Valve); ESTAD (Early-onset Sporadic Thoracic Aortic Dissection); FBAV (Familial Bicuspid Aortic Valve); TAA (Thoracic Aortic Aneurysm); TAAD (Thoracic Aortic Aneurysm & Dissection); FTAAD (Familial Thoracic Aortic Aneurysm & Dissection). Other in silico tools were used: FATHMM-MKL [Non-Coding Variants]; FATHMM-XF [Non-Coding Variants]; FATHMM [Inherited Disease – Coding Variants]. After further research some probands were found to have another variant in another causative gene: 1 USA proband in LMOD1 variant p.(Pro46Leu) [causative variant in COL3A1, c.1258G>A, p.(Gly420Ser)]; 1 USA proband in LMOD1 variant p.(Val47Leu) [causative variant in FBN1]; 18 French probands in LMOD1 variant p.(Thr295Met) [causative variant in either FBN1, MFAP5, ACTA2 or LOX], and 2 British probands [1 causative variant in FBN1 ,c.306dupC; p.(Ser104IfsTer25), & 1 possible causative variant in FBN1, c.2656C>A; p.(Pro886Thr)]; and 1 UK proband with no LMOD1 variant [causative variant FBN1: c.5110C>T, p.(Gln1704Ter)] (not shown in this table). Others were excluded after further phenotype assessment: 1 USA proband in LMOD1 variant p.(Arg156Trp) [reclassified with Moyamoya disease]. Family co-segregation of the variant was performed when proband’s family members were available: (yes) = positive co-segregation of the variant; (no) = negative co-segregation of the variant; (?) = family or families not tested; (->) = incomplete penetrance or late onset. The number of probands shown in ‘Other causative gen (exclusion)’ and ‘Variant segregated with disease in the family’ is indicated in brackets by a number before the letter ‘x’.
Table 1C: Final variant interpretation table of only likely pathogenic LMOD1 alleles. Note that two variants in black bold [p.(Val167Ala) & p.(Met562Ile)] were included in the final list because of their location in the protein and their very low allele frequency, even though the in silico tools agreed that they might be variants of unknown significance. The last column of this table shows the total number of in silico tools with damaging scores (+). The variants in this table are found in figure2 with upward lines showing their location in the protein. Interesting to notice that the variants classified as causative variants (coloured in red) are located either at the N- or C-terminus of the protein.
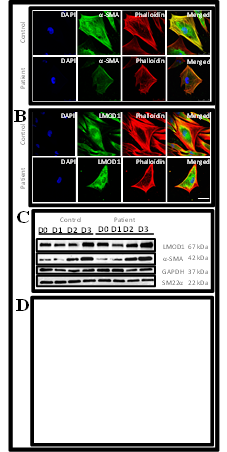
Figure 3- Representative immunofluorescence imaging of patient cells carrying the LMOD1 c.1784T>C, p.(Val595Ala) variant compared with control cells. A- Imaging of alpha-actin-2 antibody staining (a-SMA, green) at 3 days post-TGF-b1 treatment of control and patient myofibroblast cells. Less actin filaments were observed in the patient cells compared with those of controls as observed in the merged image in yellow colour. DAPI (4ʹ,6-diamidino-2-phenylindole) was used for detecting the nuclei (blue), Phalloidin was used for detecting filamentous actin (red) and the last column shows the merged image of all antibody stains used. Scale bar 50μm. Images taken at 40X magnification. B- Imaging of Leiomodin-1 antibody staining (LMOD1, green) at 3 days post-TGF-b1 treatment of control and patient myofibroblast cells. LMOD1 protein localization was in the peri-nuclear region compared with diffuse staining throughout the cytoplasm in the patient cells. DAPI (4ʹ,6-diamidino-2-phenylindole) was used for detecting the nuclei (blue), Phalloidin was used for detecting filamentous actin (red) and the last column shows the merged image of all antibody stains used. Scale bar 50μm. Images taken at 40X magnification. C- Western blot analysis of control and patient cells of LMOD1 and actin monomers (a-SMA) in the presence of housekeeping genes GAPDH and SM22a. Results showed no changes in protein concentration between patient and control cells. D- Collagen gel assay13,14 to test activity of myofibroblasts of patient and control samples (n=14 in each group) (Supplement 2). Control cell-containing collagen gels showed statistically significant reduction in size after TGFb1 treatment compared with patient cell-containing gels suggesting that the variant c.1784T>C, p.(Val595Ala) may affect the contractility of myofibroblast cells.
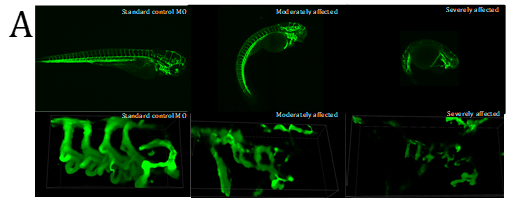
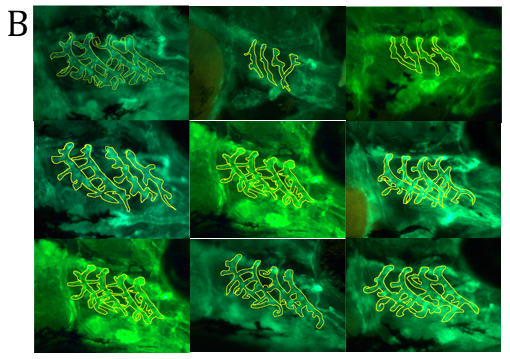
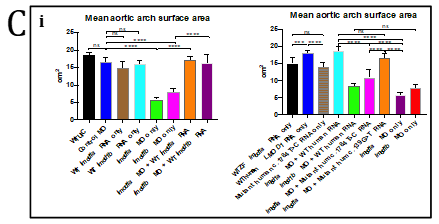
Figure 4. Fluorescent imaging of standard control Morpholino Oligonucleotide (MO) and lmod1a/lmod1b MO morphant (moderately and severely affected) kdrl:EGFP transgenic zebrafish embryos. A- Confocal imaging of whole embryo (top, scale bar- 500uM) and 3D imaging of the pharyngeal arches (bottom, scale bar- 10uM). Severely affected embryos showed incomplete vasculature throughout the embryo compared with control. The 3D image of the pharyngeal arches also show incomplete aortic arches, suggesting a delayed development. B- Epi-fluorescent imaging of pharyngeal arches. Aortic arches of standard control MO and wild-type RNA injection appear indistinguishable. lmod1a MO only and lmod1b MO only significantly reduced the surface area of the aortic arches. Rescue experiments by co-injecting lmod1a and wild-type zebrafish lmod1a RNA or lmod1b and wild-type zebrafish lmod1b RNA showed aortic arches that were similar to those of control MO. Scale bar-10uM. C- Zebrafish aortic arch surface area comparison. i- with zebrafish lmod1a and lmod1b rescue. Comparison of mean value of aortic arches surface area for each study group. No significant differences could be calculated between UIC, Control MO, WT lmod1a RNA only and WT lmod1b RNA only. Significant reduction in the surface area could be calculated between lmod1a MO only with control MO and lmod1b with control MO. Co-injection of either lmod1 MO with the respective WT lmod RNA study groups with the MO only study groups demonstrated a significant difference. However, when comparing with the Control MO, these were not significant, indicating rescue of the MO-phenotype was achieved and provided evidence of specificity of the MO. ii- comparison with human LMOD1 rescue. lmod1a co-injected with mutant human RNA containing a predicted pathogenic variant c.1784T>C demonstrated reduced aortic arch surface area when comparing with lmod1a MO with WT human RNA as well as lmod1a MO co-injected with RNA containing a predicted benign variant c.139G>T. lmod1a co-injected with mutant human RNA containing a predicted pathogenic variant c.1784T>C also demonstrated significantly larger surface area compared with lmod1a MO only injection groups, but significantly lower than WT human RNA only. ns- not significant and ****- extremely significant (p < 0.0001).
off-target effect of MO-knockdown70. Therefore, p53 MO was co-injected with both lmod1 MOs at a final concentration of 6ng to block any phenotypes associated with this off-target event. No changes were observed in the severity of the morphant phenotypes between lmod1a-b and lmod1a-b/p53 morphants. This suggests that the phenotype observed was unlikely due to the off-target effect of MOs or upregulation of p53.
Lmod1 is required for pharyngeal arch development.
An F-actin (Phalloidin) marker was used to determine if knockdown of lmod1a/lmod1b zebrafish causes alterations in the actin filament production as reported in the Lmod1-/- mouse12. A global reduction in fluorescence was observed in the trunk and tail region and incomplete pharyngeal arches were observed in the morphants. This suggested that the vasculature of lmod1-deficient embryos is impaired. A transgenic zebrafish line (kdrl:EGFP), which expresses green fluorescent protein on the surface of endothelial cells was utilized to study the vasculature in the developing pharyngeal arches. Co-injection of lmod1a/lmod1b at 4ng/ul demonstrated greatly reduced fluorescence in the posterior end of severely affected embryos, indicating fewer intersegmental vessels compared with controls (figure 4A). The pharyngeal arches in the morphants appear to develop similarly to those in the controls, but sub-branches were underdeveloped in the majority of embryos by 4dpf when normally these would have completed their branching, suggesting delay in development. 3D images of z-stacks demonstrated diffuse fluorescence in the morphants, which did not conform to the normal shape of the arches, suggesting disrupted cellular integrity.
Development of the aortic arches was studied by imaging MO-induced knockdown of kdrl:EGFP embryos between 3 to 5 days post fertilisation (dpf) using fluorescent microscopy. In the MO control group, at 3 dpf the development of the aortic arches 3-6 was complete, with sprouting of the sub-branches visible. At 4 dpf, the sub-branches elongate and begin to loop in the wild-type and remain similar until 5 dpf. In the lmod1a/lmod1b co-injection group, at 3 dpf, only aortic arches 3-4 were visible. At 4 dpf, aortic arches 5-6 have formed and at 5 dpf sprouting of the sub-branches could be seen. Together these data suggest that lmod1-deficient embryos present with delayed development of the pharyngeal arches and that lmod1 is required for the normal developing vasculature.
The global phenotype produced by co-injection of lmod1a/lmod1b at 4ng/ul concentration resembles the more severe human MMIHS phenotype rather than that of the TAAD patients in this study. Therefore, we hypothesized that at half concentration (2ng/ul) of lmod1a/lmod1b MO, we would induce the non-syndromic TAAD phenotype alone. Controls and mutants were virtually indistinguishable by light microscopy. Only minor differences could be observed under fluorescent microscopy in the pharyngeal arches of embryos. The surface area of the pharyngeal arches was measured to determine if there were statistically significant differences between study groups (figures 4B and 4C). Measurements of the standard control MO group did not deviate from the MO control group (adjusted p=0.0616), suggesting that MO at 2ng did not hinder the development of the pharyngeal arches. The lmod1a and lmod1b only study groups demonstrated a significant reduction of the surface area compared with control MO. To validate the lmod1 MOs and to test the functional relevance of the human variants, we injected human RNA to rescue and/or overexpress wild-type and mutant LMOD1 in zebrafish. Overexpression by injection of wild-type human LMOD1 RNA did not affect the development of the aortic arches when compared with MO control (adjusted p>0.9999). Comparison of lmod1a MO and human wild-type LMOD1 study group with lmod1a MO only study group demonstrated significant rescue (adjusted p<0.0001) of the pharyngeal arch phenotype. Co-injection of lmod1a MO with mutant c.1784T>C LMOD1 RNA, which is predicted to be damaging, was able to only partially rescue the lmod1a MO-phenotype when compared with lmod1a MO only study group (adjusted p=0.0048) and lmod1a MO and wild-type LMOD1 RNA study group (adjusted p<0.0001). Another mutant c.139G>T LMOD1 RNA, which is predicted to be benign was able to perform a full rescue of the lmod1a MO-phenotype when compared with lmod1a MO only study group (adjusted p<0.0001) and lmod1a MO and wild-type LMOD1 RNA study group (adjusted p=0.1801).
Discussion
The understanding of thoracic aortic aneurysm & dissection development continues to reveal novel genes that predispose individuals to TAAD such as THSD450, where the encoded protein product (ADAMTSL6) binds directly to fibrillin-1 and promotes fibrillin-1 matrix assembly71. Other studies have shown that several variants in the contractile proteins of smooth muscle cells predispose individuals to TAA4,9, suggesting that smooth muscle contractile function plays an important role in TAA though the mechanism is unclear. This is getting more noticeable when looking at the smooth muscle cells (SMC), which are predominantly in the aortic medial layer and maintain the wall structure, where downregulation of LMOD1 & MYH11 genes has been associated with bulging of the weakened blood vessels wall in intracranial aneurysm (IA) phenotype72, and also a reduce LMOD1 expression in coronary artery disease (CAD)73. Two more connective tissue phenotypes associated with SMC disfunctions. Genetic disfunction in SMC contractile proteins and their devastating consequences for the aortic wall have been shown in cases of familial thoracic aneurysm53,74.
This study introduces LMOD1 as the next critical component of the contractile apparatus in smooth muscle, since variants in the LMOD1 gene predispose to TAAD.
LMOD1
Leiomodins (LMODs) [powerful filament nucleators] and Tropomodulins (TMODs) [pointed-end capping] are a group of proteins that are involved in actin cytoskeleton assembly, and both groups can be distinguished by the absence of the TMBS2 domain, and more interestingly by the presence of a proline-rich region and a WH2 domain seen in the C-terminus extension in Lmods68,75. Leiomodins are three paralogous genes (LMOD1, LMOD2 and LMOD3). Lmod1 has a preferential expression in differentiated smooth muscle cells (SMC); while Lmod2 and Lmod3 are highly restricted to cardiac and skeletal muscles with very low levels in SMC-containing tissues76-80.
Leiomodin-1 (Lmod1) is a 64-kDa actin-binding protein that functions as a nucleator of actin monomers to filaments68. Lmod1 is expressed predominantly in visceral and vascular smooth muscle cells (SMC), with very high expression in the aorta and bladder76, but it can also be found in intestinal tissue79, arteries, gastrointestinal tract, bladder and in tissues both sexes60.
Several known structures or domains are known in Lmod1 [TMBS, ABS1, ABS2, PRR and WH2] (figure 2). The C-terminus extension in Lmod1 (true in all 3 paralogous genes) consists of a proline-rich region (PRR) followed by a WH2 domain78. Recent studies have shown that constructs of Lmod1 lacking the C-terminus, containing proline-rich and WH2 domains, cause a delay in actin polymerization and increased dependence of LMOD1 concentration relative to the polymerization rate68. Therefore, the domains at the C-terminus could have a secondary role in their activity78, and studying variants in the WH2 domain should elucidate this secondary role. Many of the proteins known to regulate actin dynamics contain a WH2 domain, such as the β-thymosin81 family of proteins, and WASP proteins bind the actin nucleating protein complex Arp2/382. These data above suggest that the variants found in the C-terminus extension containing the actin-binding domain (WH2) of LMOD1 may delay actin polymerization and therefore compromise the length and dynamics of actin filaments. Our findings of reduced SMA and phalloidin staining in the cellular studies highlight a reduction in actin filaments in the cytoplasm, suggesting that the variant in LMOD1 may be limiting the binding and nucleation of actin filaments. Our results also support previously reported findings.
It has been studied that gene variants which lead to decreased contraction of vascular SMCs can cause inherited TAAD44,53 in heterozygote variants, and also MMIHS12 in homozygote variants. LMOD1 was previously described as causing an autosomal recessive, fatal rare condition, MMIHS, in a newborn child, and also in mice12 (see Supplement 3 for Genetic Risk Assessment for LMOD1, MYLK & MYH11 genes).
Loss of Lmod1 leads to defective actin filament assembly and reduced contractility in visceral smooth muscle12. Although Lmod1 is abundant in both vascular and visceral smooth muscles12,76,77,79, Lmod1-/- mice exhibit pathological thinning and compaction of visceral smooth muscle (stomach, bladder, intestine), but surprisingly, not vascular (aorta, oesophagus) smooth muscles12. Even though, it has been reported a twofold decrease in LMOD1 protein in stomach, aorta, and bladder of heterozygous mice, and even lower levels in compound heterozygous mice, whereas undetectable levels were observed in homozygous mice12. However, they reported that such compound heterozygous mice were without noticeable defect, and they pointed out that a low-level threshold of LMOD1 protein might exist for normal homeostasis.
siRNA knockdown of human LMOD1 in intestinal smooth muscle cells showed decrease in actin filament formation and contractility12. Lmod1-/- mouse bladder tissue displayed fewer actin filaments and rod-like structures (nemaline bodies – might cause muscle weakness83,84), similarly found in the Lmod3-/- mouse. The authors demonstrated that LMOD1 has significant importance in regulating smooth muscle cytoskeletal-contractile coupling and indicated that lack of Lmod1 actin filament nucleation in contractile structures leads to inappropriate actin assembly in noncontractile Z-line structures.
LMOD1 variants
Non-synonymous LMOD1 variants identified in this study, the American [Arg575Cys], the Dutch [Leu590Pro] and the British [Val595Ala], all result in a change in an amino acid residue located in the WH2 domain at residues 575-60068 of the protein. Also, the American [Glu22Ter] non-sense variant is expected to shorten the protein, if mRNA decay does not occur, which will delete the TMBS, ABS2 and WH2 domain completely.
The WH2 domain is variable in length and sequence but contains the conserved four residue motif LKK(T/V) [residues 580/581/582/583], which is critical for the interaction with actin85. Changes in the structure of the WH2 domain may be interfering with the binding to, and assembly of, actin filaments therefore disrupting the actin dynamics and interaction with myosin. Since WH2 domain is the 3rd Actin Binding Site it might not fully disrupt the full function of the protein but delay it, labelling the WH2 domain as an auxiliary role in nucleation68. Delayed actin polymerization by abnormal LMOD1 may lead to earlier capping of the pointed actin filaments by Tmods, resulting in shorter actin filaments.
Lmods interacts with TM (tropomyosin) protein through the TMBS (tropomodulin binding site) domain68,78,86. Tmods work in conjunction with one of several TM isoforms to cap the pointed end of actin filaments in cytoskeletal structures87 through two different sites88, and only the first site is conserved in Lmods68. The sequence of residues involved in the binding of TMBS domain in LMOD1 to TM is not fully understood, but an earlier study suggested that residues 6-94 of Tmod1 contain the binding site for skeletal muscle TM89. Full length Tmod1 with mutated residue Leu27 in the TMBS1 domain had no significant influence on the capping ability88, which in previous studies with the same mutated residue led to a reduce TM affinity and partial capping function in the presence of TM in fragments of Tmod (Tmod11-92 and Tmod11-48)90,91. The TMBS2 domain of Tmods might compensate for an abnormal TMBS1 domain, but LMOD1 might not achieve this since there is only one TMBS domain at the N-terminus, and therefore, the TMBS domain must be crucial for LMOD1 function. Our identified variant LMOD1 Arg9Trp located in the TMBS was predicted to be pathogenic from the majority of in silico tools, and it might be involved in the binding of TM.
Another variant identified in this study, Thr55Met located between the TMBS and the ABS1 (Actin binding site 1) was predicted to be pathogenic from the majority of in silico tools. We cannot classify this variant as benign since regions of unclear significance in LMOD1 gene have been shown to be imperative for its activity73, and our identified variant might contribute to the severity of the disease.
The variant Pro544His (MAF ≤0.00003) is located in the proline-rich region (PRR) considered as an actin-binding contributor92, which may account for the TAAD phenotype observed in the UK TAAD family M508, and also, in one of the Belgium-Antwerp families. It was classified as a variant of unknown significance (VUS) since it was filtered out when looking at the MAF (≤0.000007).
It is very interesting that most of our TAAD cohort variants were found in the N-terminus (19/25) with 2 predicted to be causative in the TMBS domain, and a few of them in the C-terminus (6/25) with 3 predicted to be causative in the WH2 domain. When performing burden tests on our data, we found that rare WH2 domain (MAF ≤0.00001) and TMBS domain (MAF ≤0.00001315) variants were enriched in the complete TAAD patient cohort compared with that in gnomAD (p<3.077e-15 and p<0.01121 respectively) (Table S5.3 and S5.5), and borderline significance (p<0.05063) occurred when analysing the rare variants (MAF ≤0.00001315) from the TMBS & ABS1 domain (Table S5.4), even though the allele frequency was three times higher in the TAAD population.
These data could indicate that variants found in the TMBS domain (N-terminus domain found twice in TMODs) and WH2 domain (C-terminus domain absent in TMODs) must be important for the full function of the protein and the host.
This study focused on two large LMOD1 TAAD families: one from our UK cohort (M441) and one from the Dutch cohort (Netherlands01). The incidence of LMOD1 disease-related variants in our UK cohort is ~1.1% (1/91) and the phenotypic presentation of this gene may be of late onset, as observed by the mean age of dissection in males (50.4 years, range 39-58 years) and variable expression of this dominant gene between carrier males and females in our study family (M441). Of 18 adults over the age of 18 (7M:11F) carrying the c.1784T>C allele, those six with demonstrated aneurysm were predominantly male (5M:1F). We must also say from the point of view of the Dutch family that the phenotype can arise at any time without previous warning as observed in affected patient IV:2 (figure 1B). Therefore, long term monitoring is critical for family members carrying causative LMOD1 variants who are classified as asymptomatic carriers or with ambiguous phenotypes.
LMOD1 animal model
Our c.1784T>C variant containing RNA study in zebrafish may demonstrate either a dominant negative effect or a haploinsufficient state but illustrates ‘loss of function’ mechanism like the one seen in MMIHS phenotype, as the resulting phenotype is similar to that observed in lmod1a/lmod1b ‘knockdown’ in zebrafish. Alternative concentration doses (>2-4ng/ul<) might induce the full non-syndromic TAAD phenotype alone, excluding the ‘loss of function’ effect seen in MMIHS, since the 2ng/ul could only reproduce minor differences under fluorescent microscopy in the pharyngeal arches of embryos, which might be enough to demonstrate an abnormal function of LMOD1. The pharyngeal arches in zebrafish develop to become the gills in adults for efficient uptake of oxygen, and in humans, these structures morphologically change to become part of the thoracic aorta as seen in the human. Abnormal development of the pharyngeal arches in humans lead to congenital cardiovascular deformities, such as Patent Ductus Arteriosus (PDA) often linked to pathogenic variants in the TAAD gene MYH117,11.
The likely dominant negative phenotype suggested by our animal studies indicates a delay on the full function of the variant carried by our study UK family. The variant is likely to cause a delay in the nucleation of actin monomers to actin filaments because of a non-functioning WH2 domain, resulting in decreased actin filaments12. Therefore, the crosslinking assembly of actin filaments may be impaired, as abnormal distributions and lower concentrations of phalloidin staining have been observed by us and Halim et al12, which may consequently be leading to aortic cystic medial necrosis and permitting dilatation and dissection in the affected individuals of our study family.
The abnormal branching of the aortic arches observed in the Tg(kdrl:EGFP) zebrafish may become an aneurysm specific gene phenotype as this might now be the third TAAD gene implicated in abnormal branching of the pharyngeal arches following the FOXE326 and MAT2A28 reports.
The zebrafish study with c.1784T>C variant added further evidence to suggest that this variant impairs the development of the aortic arches, and it can be classified as disease-causing variant. The zebrafish data in our study suggest that the aortic arch defect is specific to abnormal lmod1, the morpholinos specifically target their corresponding gene, and the LMOD1 function is conserved between zebrafish and humans.
Five further variants in our international cohorts were classed as likely pathogenic and further segregation studies and animal modelling are needed to confirm these. Detailed analyses performed using Lmod1-/- mice, revealed MMIHS phenotype to be a consequence of disrupted actin filament assembly, cytoskeletal arrangement and impaired SMC contractility12. Further studies at later stages in Lmod1+/- mice would have been very informative to confirm no development of MMIHS in the heterozygous state and revealed, perhaps, the TAAD phenotype, since TAAD in patients with an LMOD1 variant is expected to be developed at a later age.
LMOD1 suggested underlying mechanism to TAAD
This study showed one non-sense LMOD1 variant (Glu22Ter) in a patient showing TAAD phenotype, which is predicted by in silico tools to be deleterious, and it is predicted to have a degradation of the variant transcript via the nonsense-mediated mRNA decay pathway. These data might suggest that reduction in amounts of LMOD1, rather than the production of variant protein, accounts for the shortness of actin filaments in smooth muscle cells, consistent with a haploinsufficiency model of TAAD pathogenesis.
Furthermore, this study showed one non-synonymous LMOD1 variant (Arg9Trp) in the TMBS domain [N-terminus], and three non-synonymous LMOD1 variants (Arg575Cys, Leu590Pro, and Val595Ala) in the WH2 domain [C-terminus] that have been seen in patients showing TAAD phenotype. All four variants are predicted by in silico tools to be deleterious, and therefore, they might disrupt the normal function of their domain in the LMOD1 protein. Evidence of reduced tropomyosin (TM) affinity and partial capping function in abnormal TMBS domain90,91, evidence of reduce nucleation activity after removal of the C-terminus, which contains the WH2 domain in Lmod168, and in this study, partial rescue when injecting the c.1784T>C LMOD1 RNA, while c.139G>T LMOD1 RNA was able to perform a full rescue of the lmod1a MO-phenotype, and reduction of actin filaments in the cytoplasm of c.1784T>C myofibroblast cells that were less able to contract, could account for the shortness of actin filaments in smooth muscle cells, consistent with a dominant negative model of TAAD pathogenesis.
From this study, we would like to suggest two major underlaying mechanisms of TAAD pathogenesis from patients with heterozygous variants in LMOD1. A haploinsufficiency model that will indicate shorter than normal actin filaments in smooth muscle cells caused by reduced amount of LMOD1 to polymerise actin filaments resulting in TAAD, and a dominant negative model that will indicate shorter than normal actin filaments in smooth muscle cells caused by reduce activity of defective LMOD1 to polymerise actin filaments resulting in TAAD.
These two suggested models will have to be studied in molecular/cellular assays in more patients with LMOD1 heterozygous variants to strongly establish them as the underlying mechanisms for this type of TAAD.
Conclusion
We have shown that variants in the N-terminus & C-terminus end of LMOD1 may underlie TAAD with an international incidence of ~ 0.5%. Additional mammalian in-vivo knockdown studies may help to further confirm a role of LMOD1 in actin dynamics of the aorta. Histopathology will be required to explore the pathological effects (such as demonstration of cystic medial necrosis) of these variants on the vascular smooth muscle wall in TAAD. An associated phenotype may be established, when more families and individuals with pathogenic alleles have been found. Further research should include in vitro cell line models to quantify and clarify their expression. LMOD1 should be investigated in TAAD exome profiles already available internationally and it may be useful to add this small three-exon gene to future TAAD panels. Sequencing of other international TAAD cohorts will help determine a true worldwide incidence. In addition, screening other genes in this important actin-myosin pathway may highlight other TAAD candidate genes. This should lead to more specific collaborative therapeutic trials once enough affected LMOD1 deficient patients have been identified nationally and internationally.
Highlights
- First time evidence for a role of LMOD1 in the manifestation of TAAD.
- A variant in the LMOD1 WH2 domain demonstrated reduced nucleation of actin filaments, mislocalization of LMOD1 protein, and impaired contractility of myofibroblast cells.
- We have used zebrafish as a vertebrate model of disease and described a novel in vivo assay for detecting changes in the development of the pharyngeal arches (that later form part of the thoracic aorta) as a result of defective Lmod1.
Acknowledgements
The authors would like to thank the families participating in this research study and their referring physicians.
Sources of funding
This work was supported with grants from the British Heart Foundation (“PG/14/77/31089 Exome sequencing to discover new causative genes for TAAD-Thoracic Aortic Aneurysm and Dissection)”to E.R.B, The Rosetrees Trust to Y.B.A.W. and A.C, The Marfan Trust, The Marfan Association and The Everett Roseborough Legacy to Y.B.A.W, Peter and Sonia Field Charitable Trust to A.C; St George’s Medical School and NHS Hospital Trust to A.C.; Guy’s and St Thomas’s charity to A.S; and Agence Nationale pour la Recherche ANR-14-CE15-0012 to G.J. The zebrafish study was conducted in collaboration with Zebsolutions.
Conflict of interest statement
DISCLOSURE-None
Abbreviations
3D – Three-dimensional space
ABS1 – Actin binding site 1
ABS2 – Actin binding site 2
IA – Intracranial aneurysm
CAD – Coronary artery disease
CNV – Copy number variation
dfp – days post fertilisation
ESTAD – Early-onset sporadic thoracic aortic dissection
ExAc – The exome aggregation consortium
FTAAD – Familial thoracic aortic aneurysm and dissection
gnomAD – The genome aggregation database
LMOD1 – Leiomodin-1
LMODs – Leiomodins
LOD – Logarithm of the odds
MAF – Minor allele frequency
MMIHS – Megacystis microcolon intestinal hypoperistalsis syndrome
MO – Morpholino oligonucleotide
MYH11 – Myosin heavy chain 11
MYLK – Myosin light chain kinase
TAA – Thoracic aortic aneurysm
TAAD – Thoracic aortic aneurysm and dissection
TM – Tropomyosin
TMBS – Tropomodulin binding site
TMODs – Tropomodulins
SMC – Smooth muscle cells
UK – United Kingdom
USA – United States of America
VCF – Variant call format
WES – Whole exome sequencing
WH2 – Wiskott-Aldrich homology 2
References
1. Halim D, Brosens E, Muller F, et al. Loss-of-Function Variants in MYLK Cause Recessive Megacystis Microcolon Intestinal Hypoperistalsis Syndrome. Am J Hum Genet. Jul 2017;101(1):123-129. doi:10.1016/j.ajhg.2017.05.011
2. Wang L, Guo DC, Cao J, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. Nov 2010;87(5):701-7. doi:10.1016/j.ajhg.2010.10.006
3. Shalata A, Mahroom M, Milewicz DM, et al. Fatal thoracic aortic aneurysm and dissection in a large family with a novel MYLK gene mutation: delineation of the clinical phenotype. Orphanet J Rare Dis. 03 2018;13(1):41. doi:10.1186/s13023-018-0769-7
4. Wallace SE, Regalado ES, Gong L, et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet Med. 01 2019;21(1):144-151. doi:10.1038/s41436-018-0038-0
5. Wang Q, Zhang J, Wang H, Feng Q, Luo F, Xie J. Compound heterozygous variants in MYH11 underlie autosomal recessive megacystis-microcolon-intestinal hypoperistalsis syndrome in a Chinese family. J Hum Genet. Nov 2019;64(11):1067-1073. doi:10.1038/s10038-019-0651-z
6. Kloth K, Renner S, Burmester G, et al. 16p13.11 microdeletion uncovers loss-of-function of a MYH11 missense variant in a patient with megacystis-microcolon-intestinal-hypoperistalsis syndrome. Clin Genet. 07 2019;96(1):85-90. doi:10.1111/cge.13557
7. Zhu L, Vranckx R, Khau Van Kien P, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. Mar 2006;38(3):343-9. doi:10.1038/ng1721
8. Gauthier J, Ouled Amar Bencheikh B, Hamdan FF, et al. A homozygous loss-of-function variant in MYH11 in a case with megacystis-microcolon-intestinal hypoperistalsis syndrome. Eur J Hum Genet. Sep 2015;23(9):1266-8. doi:10.1038/ejhg.2014.256
9. Keravnou A, Bashiardes E, Michailidou K, Soteriou M, Moushi A, Cariolou M. Novel variants in the ACTA2 and MYH11 genes in a Cypriot family with thoracic aortic aneurysms: a case report. BMC Med Genet. 12 2018;19(1):208. doi:10.1186/s12881-018-0728-0
10. Takeda N, Morita H, Fujita D, et al. A deleterious MYH11 mutation causing familial thoracic aortic dissection. Hum Genome Var. 2015;2:15028. doi:10.1038/hgv.2015.28
11. Pannu H, Tran-Fadulu V, Papke CL, et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet. Oct 2007;16(20):2453-62. doi:ddm201 [pii]
10.1093/hmg/ddm201
12. Halim D, Wilson MP, Oliver D, et al. Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc Natl Acad Sci U S A. 03 2017;114(13):E2739-E2747. doi:10.1073/pnas.1620507114
13. Kwartler CS, Chen J, Thakur D, et al. Overexpression of smooth muscle myosin heavy chain leads to activation of the unfolded protein response and autophagic turnover of thick filament-associated proteins in vascular smooth muscle cells. J Biol Chem. May 2014;289(20):14075-88. doi:10.1074/jbc.M113.499277
14. Ngo P, Ramalingam P, Phillips JA, Furuta GT. Collagen gel contraction assay. Methods Mol Biol. 2006;341:103-9. doi:10.1385/1-59745-113-4:103
15. Hannuksela M, Stattin EL, Klar J, et al. A novel variant in MYLK causes thoracic aortic dissections: genotypic and phenotypic description. BMC Med Genet. 09 01 2016;17(1):61. doi:10.1186/s12881-016-0326-y
16. Landenhed M, Engström G, Gottsäter A, et al. Risk profiles for aortic dissection and ruptured or surgically treated aneurysms: a prospective cohort study. J Am Heart Assoc. Jan 2015;4(1):e001513. doi:10.1161/JAHA.114.001513
17. Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD, Tejada-Vera B. Deaths: final data for 2006. Natl Vital Stat Rep. Apr 2009;57(14):1-134.
18. Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. Mar 1997;25(3):506-11.
19. Coady MA, Davies RR, Roberts M, et al. Familial patterns of thoracic aortic aneurysms. Arch Surg. Apr 1999;134(4):361-7.
20. Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections–incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. Oct 2006;82(4):1400-5. doi:10.1016/j.athoracsur.2006.04.098
21. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. Jul 2010;47(7):476-85. doi:10.1136/jmg.2009.072785
22. Pyeritz RE. Heritable thoracic aortic disorders. Curr Opin Cardiol. Jan 2014;29(1):97-102. doi:10.1097/HCO.0000000000000023
23. Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. Dec 2007;39(12):1488-93. doi:10.1038/ng.2007.6
24. Guo DC, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. May 2009;84(5):617-27. doi:10.1016/j.ajhg.2009.04.007
25. Meester JA, Vandeweyer G, Pintelon I, et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet Med. Sep 2016;doi:10.1038/gim.2016.126
26. Kuang SQ, Medina-Martinez O, Guo DC, et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Invest. Mar 2016;126(3):948-61. doi:10.1172/JCI83778
27. Vermeer AMC, Lodder EM, Thomas D, et al. Dilation of the Aorta Ascendens Forms Part of the Clinical Spectrum of HCN4 Mutations. J Am Coll Cardiol. May 2016;67(19):2313-2315. doi:10.1016/j.jacc.2016.01.086
28. Guo DC, Gong L, Regalado ES, et al. MAT2A Mutations Predispose Individuals to Thoracic Aortic Aneurysms. Am J Hum Genet. Jan 2015;96(1):170-7. doi:10.1016/j.ajhg.2014.11.015
29. Barbier M, Gross MS, Aubart M, et al. MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet. Dec 2014;95(6):736-43. doi:10.1016/j.ajhg.2014.10.018
30. Micha D, Guo DC, Hilhorst-Hofstee Y, et al. SMAD2 Mutations Are Associated with Arterial Aneurysms and Dissections. Hum Mutat. Aug 2015;doi:10.1002/humu.22854
31. Bertoli-Avella AM, Gillis E, Morisaki H, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. Apr 2015;65(13):1324-36. doi:10.1016/j.jacc.2015.01.040
32. Hucthagowder V, Sausgruber N, Kim KH, Angle B, Marmorstein LY, Urban Z. Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am J Hum Genet. Jun 2006;78(6):1075-80. doi:10.1086/504304
33. Hadj-Rabia S, Callewaert BL, Bourrat E, et al. Twenty patients including 7 probands with autosomal dominant cutis laxa confirm clinical and molecular homogeneity. Orphanet J Rare Dis. Feb 2013;8:36. doi:10.1186/1750-1172-8-36
34. Park ES, Putnam EA, Chitayat D, Child A, Milewicz DM. Clustering of FBN2 mutations in patients with congenital contractural arachnodactyly indicates an important role of the domains encoded by exons 24 through 34 during human development. Am J Med Genet. Jul 1998;78(4):350-5.
35. Reinstein E, Frentz S, Morgan T, et al. Vascular and connective tissue anomalies associated with X-linked periventricular heterotopia due to mutations in Filamin A. Eur J Hum Genet. May 2013;21(5):494-502. doi:10.1038/ejhg.2012.209
36. Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. Sep 2005;437(7056):270-4. doi:10.1038/nature03940
37. Callewaert BL, Willaert A, Kerstjens-Frederikse WS, et al. Arterial tortuosity syndrome: clinical and molecular findings in 12 newly identified families. Hum Mutat. Jan 2008;29(1):150-8. doi:10.1002/humu.20623
38. Andrabi S, Bekheirnia MR, Robbins-Furman P, Lewis RA, Prior TW, Potocki L. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am J Med Genet A. May 2011;155A(5):1165-9. doi:10.1002/ajmg.a.33968
39. Doyle AJ, Doyle JJ, Bessling SL, et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. Nov 2012;44(11):1249-54. doi:10.1038/ng.2421
40. Kashtan CE, Segal Y, Flinter F, Makanjuola D, Gan JS, Watnick T. Aortic abnormalities in males with Alport syndrome. Nephrol Dial Transplant. Nov 2010;25(11):3554-60. doi:10.1093/ndt/gfq271
41. Peczkowska M, Januszewicz A, Grzeszczak W, et al. The coexistence of acute aortic dissection with autosomal dominant polycystic kidney disease–description of two hypertensive patients. Blood Press. 2004;13(5):283-6. doi:10.1080/08037050410016492
42. Superti-Furga A, Steinmann B, Ramirez F, Byers PH. Molecular defects of type III procollagen in Ehlers-Danlos syndrome type IV. Hum Genet. May 1989;82(2):104-8.
43. Guo DC, Regalado ES, Gong L, et al. LOX Mutations Predispose to Thoracic Aortic Aneurysms and Dissections. Circ Res. Mar 2016;118(6):928-34. doi:10.1161/CIRCRESAHA.115.307130
44. Guo DC, Regalado E, Casteel DE, et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. Aug 2013;93(2):398-404. doi:10.1016/j.ajhg.2013.06.019
45. Regalado ES, Guo DC, Villamizar C, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. Sep 2011;109(6):680-6. doi:10.1161/CIRCRESAHA.111.248161
46. Boileau C, Guo DC, Hanna N, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. Jul 2012;44(8):916-21. doi:10.1038/ng.2348
47. Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. Mar 2005;37(3):275-81. doi:10.1038/ng1511
48. Arnaud P, Racine C, Hanna N, et al. A new mutational hotspot in the SKI gene in the context of MFS/TAA molecular diagnosis. Hum Genet. Apr 2020;139(4):461-472. doi:10.1007/s00439-019-02102-9
49. Engström K, Vánky F, Rehnberg M, et al. Novel SMAD3 p.Arg386Thr genetic variant co-segregating with thoracic aortic aneurysm and dissection. Mol Genet Genomic Med. Apr 2020;8(4):e1089. doi:10.1002/mgg3.1089
50. Elbitar S, Renard M, Arnaud P, et al. Pathogenic variants in THSD4, encoding the ADAMTS-like 6 protein, predispose to inherited thoracic aortic aneurysm. Genet Med. 01 2021;23(1):111-122. doi:10.1038/s41436-020-00947-4
51. Dietz HC. TGF-beta in the pathogenesis and prevention of disease: a matter of aneurysmic proportions. J Clin Invest. Feb 2010;120(2):403-7. doi:10.1172/JCI42014
52. Quintana RA, Taylor WR. Cellular Mechanisms of Aortic Aneurysm Formation. Circ Res. 02 2019;124(4):607-618. doi:10.1161/CIRCRESAHA.118.313187
53. Milewicz DM, Trybus KM, Guo DC, et al. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler Thromb Vasc Biol. 01 2017;37(1):26-34. doi:10.1161/ATVBAHA.116.303229
54. Takeichi T, Liu L, Fong K, et al. Whole-exome sequencing improves mutation detection in a diagnostic epidermolysis bullosa laboratory. Br J Dermatol. Jan 2015;172(1):94-100. doi:10.1111/bjd.13190
55. Milewicz DM, Regalado ES, Shendure J, Nickerson DA, Guo DC. Successes and challenges of using whole exome sequencing to identify novel genes underlying an inherited predisposition for thoracic aortic aneurysms and acute aortic dissections. Trends Cardiovasc Med. Feb 2014;24(2):53-60. doi:10.1016/j.tcm.2013.06.004
56. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. May 2015;17(5):405-24. doi:10.1038/gim.2015.30
57. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 05 2020;581(7809):434-443. doi:10.1038/s41586-020-2308-7
58. Pundir S, Martin MJ, O’Donovan C. UniProt Protein Knowledgebase. Methods Mol Biol. 2017;1558:41-55. doi:10.1007/978-1-4939-6783-4_2
59. Consortium G. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. May 2015;348(6235):648-60. doi:10.1126/science.1262110
60. Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science. Jan 2015;347(6220):1260419. doi:10.1126/science.1260419
61. Thul PJ, Åkesson L, Wiking M, et al. A subcellular map of the human proteome. Science. 05 2017;356(6340)doi:10.1126/science.aal3321
62. Westerfield M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 5th Edition. vol 5. 2007.
63. Erbel R, Aboyans V, Boileau C, et al. [2014 ESC Guidelines on the diagnosis and treatment of aortic diseases]. Kardiol Pol. 2014;72(12):1169-252. doi:10.5603/KP.2014.0225
64. Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology,American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons,and Society for Vascular Medicine. J Am Coll Cardiol. Apr 2010;55(14):e27-e129. doi:10.1016/j.jacc.2010.02.015
65. Renard M, Francis C, Ghosh R, et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J Am Coll Cardiol. 08 2018;72(6):605-615. doi:10.1016/j.jacc.2018.04.089
66. Guo DC, Regalado ES, Pinard A, et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am J Hum Genet. 04 2018;102(4):706-712. doi:10.1016/j.ajhg.2018.03.002
67. Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi:10.1186/1471-2105-11-548
68. Boczkowska M, Rebowski G, Kremneva E, Lappalainen P, Dominguez R. How Leiomodin and Tropomodulin use a common fold for different actin assembly functions. Nat Commun. Sep 2015;6:8314. doi:10.1038/ncomms9314
69. Wittbrodt J, Meyer A, Schartl M. More genes in fish? BioEssays. 1998;20(6):511-515.
70. Robu ME, Larson JD, Nasevicius A, et al. p53 activation by knockdown technologies. PLoS Genet. May 2007;3(5):e78. doi:10.1371/journal.pgen.0030078
71. Tsutsui K, Manabe R, Yamada T, et al. ADAMTSL-6 is a novel extracellular matrix protein that binds to fibrillin-1 and promotes fibrillin-1 fibril formation. J Biol Chem. Feb 12 2010;285(7):4870-82. doi:10.1074/jbc.M109.076919
72. Cheng Q, Li Z, Wang R, et al. Genetic Profiles Related to Pathogenesis in Sporadic Intracranial Aneurysm Patients. World Neurosurg. Nov 2019;131:e23-e31. doi:10.1016/j.wneu.2019.06.110
73. Nanda V, Wang T, Pjanic M, et al. Functional regulatory mechanism of smooth muscle cell-restricted LMOD1 coronary artery disease locus. PLoS Genet. 11 2018;14(11):e1007755. doi:10.1371/journal.pgen.1007755
74. Milewicz DM, Guo DC, Tran-Fadulu V, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283-302. doi:10.1146/annurev.genom.8.080706.092303 [doi]
75. Qualmann B, Kessels MM. New players in actin polymerization–WH2-domain-containing actin nucleators. Trends Cell Biol. Jun 2009;19(6):276-85. doi:10.1016/j.tcb.2009.03.004
76. Conley CA, Fritz-Six KL, Almenar-Queralt A, Fowler VM. Leiomodins: larger members of the tropomodulin (Tmod) gene family. Genomics. Apr 2001;73(2):127-39. doi:10.1006/geno.2000.6501
77. Conley CA. Leiomodin and tropomodulin in smooth muscle. Am J Physiol Cell Physiol. Jun 2001;280(6):C1645-56. doi:10.1152/ajpcell.2001.280.6.C1645
78. Chereau D, Boczkowska M, Skwarek-Maruszewska A, et al. Leiomodin is an actin filament nucleator in muscle cells. Science. Apr 2008;320(5873):239-43. doi:10.1126/science.1155313
79. Nanda V, Miano JM. Leiomodin 1, a new serum response factor-dependent target gene expressed preferentially in differentiated smooth muscle cells. J Biol Chem. Jan 2012;287(4):2459-67. doi:10.1074/jbc.M111.302224
80. Yuen M, Sandaradura SA, Dowling JJ, et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest. Nov 2014;124(11):4693-708. doi:10.1172/JCI75199
81. Hertzog M, van Heijenoort C, Didry D, et al. The beta-thymosin/WH2 domain; structural basis for the switch from inhibition to promotion of actin assembly. Cell. May 2004;117(5):611-23.
82. Zalevsky J, Lempert L, Kranitz H, Mullins RD. Different WASP family proteins stimulate different Arp2/3 complex-dependent actin-nucleating activities. Curr Biol. Dec 2001;11(24):1903-13.
83. SHY GM, ENGEL WK, SOMERS JE, WANKO T. NEMALINE MYOPATHY. A NEW CONGENITAL MYOPATHY. Brain. Dec 1963;86:793-810. doi:10.1093/brain/86.4.793
84. Wallgren-Pettersson C, Sewry CA, Nowak KJ, Laing NG. Nemaline myopathies. Semin Pediatr Neurol. Dec 2011;18(4):230-8. doi:10.1016/j.spen.2011.10.004
85. Chereau D, Kerff F, Graceffa P, Grabarek Z, Langsetmo K, Dominguez R. Actin-bound structures of Wiskott-Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc Natl Acad Sci U S A. Nov 15 2005;102(46):16644-9. doi:10.1073/pnas.0507021102
86. Rao JN, Madasu Y, Dominguez R. Mechanism of actin filament pointed-end capping by tropomodulin. Science. Jul 25 2014;345(6195):463-7. doi:10.1126/science.1256159
87. Yamashiro S, Gokhin DS, Kimura S, Nowak RB, Fowler VM. Tropomodulins: pointed-end capping proteins that regulate actin filament architecture in diverse cell types. Cytoskeleton (Hoboken). Jun 2012;69(6):337-70. doi:10.1002/cm.21031
88. Kostyukova AS, Choy A, Rapp BA. Tropomodulin binds two tropomyosins: a novel model for actin filament capping. Biochemistry. Oct 03 2006;45(39):12068-75. doi:10.1021/bi060899i
89. Babcock GG, Fowler VM. Isoform-specific interaction of tropomodulin with skeletal muscle and erythrocyte tropomyosins. J Biol Chem. Nov 04 1994;269(44):27510-8.
90. Greenfield NJ, Kostyukova AS, Hitchcock-DeGregori SE. Structure and tropomyosin binding properties of the N-terminal capping domain of tropomodulin 1. Biophys J. Jan 2005;88(1):372-83. doi:10.1529/biophysj.104.051128
91. Kostyukova AS, Rapp BA, Choy A, Greenfield NJ, Hitchcock-DeGregori SE. Structural requirements of tropomodulin for tropomyosin binding and actin filament capping. Biochemistry. Mar 29 2005;44(12):4905-10. doi:10.1021/bi047468p
92. Urbanek AN, Smith AP, Allwood EG, Booth WI, Ayscough KR. A novel actin-binding motif in Las17/WASP nucleates actin filaments independently of Arp2/3. Curr Biol. Feb 2013;23(3):196-203. doi:10.1016/j.cub.2012.12.024
Supplement 1
Knockdown of lmod1a/lmod1b
Knockdown was achieved by use of antisense morpholino oligonucleotides (Gene Tools, Oregon, USA) targeting the splice site:
- lmod1a
- exon1/intron1 (5’-TATCATCATCATCACCTCTGCGCTC-3’)
- intron1/exon2 (5’-CCTCCCTACAAACAGACACATTATT-3’)
- lmod1b
- exon2/intron2 (5’- ATTTCCATTTCCTACCTTTTTGAGC-3’)
Supplement 2
Collagen Gel Assay
In vitro Studies protocol13,14:
Skin fibroblasts from proband (Family M441 IV:5 – see figure 1A) and age and sex matched control full thickness forearm skin biopsies were explanted and cultured in 10% FBS (Fetal Bovine Serum) DMEM (Dulbecco’s Modified Eagle’s Medium) media. Each experiment used same cell passage for control and patient cells and was performed at least three independent times at different passages (between P2 and P6). For each experiment, after reaching confluence, cells were seeded and serum starved in 1% FBS DMEM media for 24h prior to the start of the experiment, then fibroblasts were stimulated with 2ng/ml of TGFb1 for 72h to be differentiated into myofibroblasts.
For immunofluorescence studies, 5,000 cells were seeded on coverslip in 6-well plate, fixed in 4% paraformaldehyde during 15 min and blocked with a 1% BSA – 0.5% Tween 20 solution. Immunostaining was performed with a-SMA (mouse monoclonal, A5228, Sigma-Aldrich, 1/100) and LMOD1 (rabbit polyclonal, 15117-1-AP, Proteintech, 1/25) as primary antibodies and followed by secondary goat anti-Mouse IgG Alexa Fluor 488 (A-11001, Invitrogen, 1/200) for a-SMA and goat anti-Rabbit IgG Alexa Fluor 488 (A-11008, Invitrogen, 1/200) for LMOD1 and co-stained with Texas RedTM -X Phalloidin (T7471, Invitrogen, 1/40) and DAPI (included in the Vectashield mounting medium). Images were obtained from a confocal inverted microscope DMi8 from Leica.
For Immunoblotting, 150,000 cells were seeded in 6-wells plate. Proteins were isolated in RIPA buffer containing phosphatase inhibitors, sonicated for 20 seconds and the concentration was determined by Bradford assay. Samples were boiled for 5 minutes and 5μg of proteins for each sample was separated by SDS-PAGE, transferred to PVDF membrane, and probed with the following antibodies: a-SMA (mouse monoclonal, A5228, Sigma-Aldrich, 1/1,000) followed by secondary mouse HRP (1/2,500); LMOD1 (rabbit polyclonal, 15117-1-AP, Proteintech 1/700) followed by secondary rabbit HRP (1/2,500); SM-22a (rabbit ab14106, Abcam, 1/1,000) followed by secondary rabbit HRP (1/2,500); and GAPDH (rabbit mAB 14C10, #2118s, Cell Signaling Technology 1/10,000) followed by secondary rabbit HRP (1/2,500) following standard protocol.
Collagen gel assays were used to assess the contractility of patient versus control myofibroblasts following a published protocol13,14. Cells were separated int 100,000 cell aliquots and seeded within a matrix of type I collagen from rat tail (BD Biosciences) into 24-well plate. The collagen mixture was prepared according to the manufacturer’s specifications at a final concentration of 1 mg/ml of collagen. Cells were resuspended into the collagen and seeded into the wells, and gels were allowed to polymerize at room temperature for at least 1h, then 1% FBS DMEM media was immediately added. Next, 24h later, TGFb1 was added to the media and gels were allowed to incubate at 37oC 5% CO2 for 3 days to accumulate tension, and then released from attachment to the cell culture plastic to induce contraction. Gels were photographed 30 min after release, and measurements were performed using ImageJ software. Several pictures were taken to quantify the staining signal.
Supplement 3
Genetic Risk Assessment for MYLK, MYH11 & LMOD1 genes
Figure S3.1: Flowchart for the genetic risk assessment for causative variants in MYLK. FTAAD (Familial Thoracic Aortic Aneurysm & Dissection). MMIHS1 (Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome Type 1). *Published data.
Symbols: causative variants observed not observed.
MYLK – Genetic Risk Assessment
Figure S3.2: Flowchart for the genetic risk assessment for causative variants in MYH11. FTAAD (Familial Thoracic Aortic Aneurysm & Dissection). MMIHS2 (Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome Type 2). *Published data. Symbols: causative variants observed and not observed in the gene.
MYH11 – Genetic Risk Assessment
Figure S3.3: Flowchart for the genetic risk assessment for causative variants in LMOD1. FTAAD (Familial Thoracic Aortic Aneurysm & Dissection). MMIHS3 (Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome Type 3). *Published data. #This study. Symbols: causative variants observed and not observed in the gene.
LMOD1 – Genetic Risk Assessment

Supplement 4
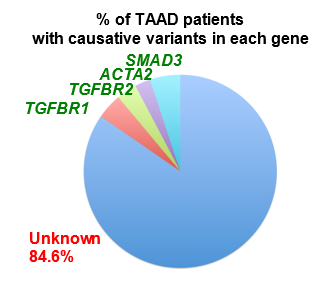
Figure S4.1: Percentage of TAAD patients explained by published causative genes in our study population TGFBR1 (4.3%), TGFBR2 (3.4%), ACTA2 (2.6%) and SMAD3 (5.1%). There are 84.6% that remain unexplained in our present study cohort.
Sonalee Laboratory TAAD Cohort
We have recruited 117 operated probands, who do not have a fibrillin-1 causative variant (previously screened by either SSCA, dHPLC or Sanger sequencing) and have a family history of aortic aneurysm. Ehlers-Danlos vascular subtype IV has been excluded clinically, by physical examination and family history, by an experienced cardiovascular geneticist (Dr A. Child).
In this cohort of 117 operated probands, we have undertaken screening (Sanger sequencing [only coding regions]) of genes previously reported and have identified pathogenic variants in 18 of the 117 (15.4%) probands, including SMAD3 in 6/117 (5.1%), ACTA2 in 3/117 (2.6%), TGFBR2 in 4/117 (3.4%), and TGFBR1 in 5/117 (4.3%). There are 99 probands (84.6%) that remain unexplained in our present study cohort.
Overall, our TAAD cohort was made of 99 probands (figure S4.2 in Supplement 4):
54 multiplex families
- 21 large families – 2-4 generations
- 33 small families
45 sporadic probands
Figure S4.2: Flow diagram of the TAAD families in this study. Initial TAAD cohort of 117 probands, where 18 had a causative gene and 99 were included in this study.
Supplement 5
- Burden test (all TAAD cohort)
A “burden test” was done with the “all TAAD cohort” in LMOD1 using a public genomic database (gnomAD v3.1.2) in RStudio (v1.4.1717 [R version = R4.1.1]) with the following result: [X2 (1, N = 202186) = 876.8, p<1.08e-192].
Data and the calculations
Total of 25 variants in LMOD1 gene were found in 93 alleles (all were heterozygote variants except Thr295Met):
21/90 alleles in France
51/179 alleles in UK
1/88 alleles in Belgium-Ghent
5/1580 alleles in Belgium-Antwerp
14/1560 alleles in USA
1/2 alleles in Netherlands
Variants were classified as:
1 synonymous
1 nonsense
22 non-synonymous
1 inframe_deletion
Total Probands: 1749.5 (1660 + 89.5) probands:
45 France
89.5 UK (this is an average)
44 Belgium-Ghent
790 Belgium-Antwerp
780 USA
1 Netherlands
The UK TAAD cohort allele final number (179) is an average from these variants:
Va47Leu – observed 1/182 alleles (91 probands)
Gly142Asp – observed 1/182 alleles (91 probands)
Thr185= – observed 1/182 alleles (91 probands)
Thr295Met – observed 46/166 alleles (83 probands) (TT = 5 / CT = 36 / CC= 42 – probands)
Prp544His – observed 1/182 alleles (91 probands)
Val595Ala – observed 1/180 alleles (90 probands)
Total alleles: 3499 alleles
International TAAD cohort 1660 * 2 = 3320 alleles
UK TAAD cohort 89.5 * 2 = 179 alleles
Genetic Database:
gnomAD (v3.1.2) all
Variant – Exclusion Criteria:
3_prime_UTR_variant
5_prime_UTR_variant
intron_variant
splice_region_variant
frameshift_variant
start_lost
Variant – Inclusion Criteria:
synonymous_variant
stop_gained = nonsense
missense_variant = non-synonymous
inframe_deletion
Formula and command line for RStudio
>LMOD1_ChiSq_test <- matrix(c(93,3499,46984,151610),ncol=2,byrow=TRUE); colnames(LMOD1_ChiSq_test) <- c(“Yes_LMOD1_variants”,”No_LMOD1_variants”); rownames(LMOD1_ChiSq_test) <- c(“TAAD”,”gnomAD_v3.1.2_All”); LMOD1_ChiSq_test <- as.table(LMOD1_ChiSq_test); LMOD1_ChiSq_test; chisq <- chisq.test(LMOD1_ChiSq_test); chisq; expected <- as.array(margin.table(LMOD1_ChiSq_test,1)) %*% t(as.array(margin.table(LMOD1_ChiSq_test,2))) / margin.table(LMOD1_ChiSq_test); expected; chi <- sum((expected – as.array(LMOD1_ChiSq_test))^2/expected); chi; pchisq(chi,df=1,lower.tail = FALSE); ft <- fisher.test(LMOD1_ChiSq_test); ft; summary(LMOD1_ChiSq_test)
| Table S5.1 ChiSq test – LMOD1 (gnomAD v3.1.2) All TAAD cohort | |||
| Yes LMOD1 | No LMOD1 | Allele frequency | |
| TAAD (All) | 93 | 3499 | 0.03 |
| gnomAD (All) | 46984 | 151610 | 0.31 |
Result: X2 (1, N = 202186) = 876.8, p<1.08e-192)
- Burden test (only UK TAAD cohort)
A “burden test” was done with the UK TAAD cohort in LMOD1 with a public genomic database (gnomAD v3.1.2 – European [non-Finnish]) using RStudio (v1.4.1717 [R version = R4.1.1]) with the following result: [X2 (1, N = 90968) = 1.1179, p<0.2904].
Data and the calculations
Total of 6 variants in LMOD1 gene were found in 51 alleles (all were heterozygote variants except Thr295Met):
Va47Leu – observed 1/182 alleles (91 probands)
Gly142Asp – observed 1/182 alleles (91 probands)
Thr185= – observed 1/182 alleles (91 probands)
Thr295Met – observed 46/166 alleles (83 probands) (TT = 5 / CT = 36 / CC= 42 – probands)
Prp544His – observed 1/182 alleles (91 probands)
Val595Ala – observed 1/180 alleles (90 probands)
Variants were classified as:
1 synonymous
5 non-synonymous
Total UK probands:
89.5 probands (this is an average)
Total alleles: 179 alleles
UK TAAD cohort 89.5 * 2 = 179 alleles
Genetic Database:
gnomAD (v3.1.2) all
Variant – Exclusion Criteria:
3_prime_UTR_variant
5_prime_UTR_variant
intron_variant
splice_region_variant
frameshift_variant
start_lost
Variant – Inclusion Criteria:
synonymous_variant
stop_gained = nonsense
missense_variant = non-synonymous
inframe_deletion
Formula for the RStudio
>LMOD1_ChiSq_test <- matrix(c(51,179,22870,67868),ncol=2,byrow=TRUE); colnames(LMOD1_ChiSq_test) <- c(“Yes_LMOD1_variants”,”No_LMOD1_variants”); rownames(LMOD1_ChiSq_test) <- c(“TAAD”,”gnomAD_v3.1.2_European_[non-Finnish]”); LMOD1_ChiSq_test <- as.table(LMOD1_ChiSq_test); LMOD1_ChiSq_test; chisq <- chisq.test(LMOD1_ChiSq_test); chisq; expected <- as.array(margin.table(LMOD1_ChiSq_test,1)) %*% t(as.array(margin.table(LMOD1_ChiSq_test,2))) / margin.table(LMOD1_ChiSq_test); expected; chi <- sum((expected – as.array(LMOD1_ChiSq_test))^2/expected); chi; pchisq(chi,df=1,lower.tail = FALSE); ft <- fisher.test(LMOD1_ChiSq_test); ft; summary(LMOD1_ChiSq_test)
| Table S5.2 ChiSq test – LMOD1 (gnomAD v3.1.2) Only UK TAAD cohort | |||
| Yes LMOD1 | No LMOD1 | Allele frequency | |
| TAAD (only UK Cohort) | 51 | 179 | 0.29 |
| gnomAD [European (non-Finnish)] | 22870 | 67868 | 0.34 |
Result: X2 (1, N = 90968) = 1.1179, p<0.2904)
- Burden test (all TAAD cohort in WH2 domain)
A “burden test” was done with the “all TAAD cohort” with only the variants found in the WH2 domain of the LMOD1 gene using a public genomic database (gnomAD v3.1.2) in RStudio (v1.4.1717 [R version = R4.1.1]) with the following result: [X2 (1, N = 155675) = 62.22, p<3.077e-15].
Data and the calculations
Total of 3 variants in the WH2 domain of the LMOD1 gene were found in 3 alleles (all were heterozygotes):
0/90 alleles in France
1/179 alleles in UK
0/88 alleles in Belgium-Ghent
0/1580 alleles in Belgium-Antwerp
1/1560 alleles in USA
1/2 alleles in Netherlands
Variants were classified as:
3 non-synonymous
Total Probands: 1749.5 (1660 + 89.5) probands:
45 France
89.5 UK (this is an average)
44 Belgium-Ghent
790 Belgium-Antwerp
780 USA
1 Netherlands
The UK TAAD cohort allele final number (179) is an average from these variants:
Va47Leu – observed 1/182 alleles (91 probands)
Gly142Asp – observed 1/182 alleles (91 probands)
Thr185= – observed 1/182 alleles (91 probands)
Thr295Met – observed 46/166 alleles (83 probands) (TT = 5 / CT = 36 / CC= 42 – probands)
Prp544His – observed 1/182 alleles (91 probands)
Val595Ala – observed 1/180 alleles (90 probands)
Total alleles: 3499 alleles
International TAAD cohort 1660 * 2 = 3320 alleles
UK TAAD cohort 89.5 * 2 = 179 alleles
Genetic Database:
gnomAD (v3.1.2) all
Variant – Exclusion Criteria: none
Variant – Inclusion Criteria:
Variant found in the WH2 domain (residues 575-600 of the protein)
MAF ≤0.00001315
Formula and command line for RStudio
>LMOD1_ChiSq_test <- matrix(c(3,3499,3,152170),ncol=2,byrow=TRUE); colnames(LMOD1_ChiSq_test) <- c(“Yes_LMOD1_variants”,”No_LMOD1_variants”); rownames(LMOD1_ChiSq_test) <- c(“TAAD_WH2″,”gnomAD_WH2”); LMOD1_ChiSq_test <- as.table(LMOD1_ChiSq_test); LMOD1_ChiSq_test; chisq <- chisq.test(LMOD1_ChiSq_test); chisq; expected <- as.array(margin.table(LMOD1_ChiSq_test,1)) %*% t(as.array(margin.table(LMOD1_ChiSq_test,2))) / margin.table(LMOD1_ChiSq_test); expected; chi <- sum((expected – as.array(LMOD1_ChiSq_test))^2/expected); chi; pchisq(chi,df=1,lower.tail = FALSE); ft <- fisher.test(LMOD1_ChiSq_test); ft; summary(LMOD1_ChiSq_test)
| Table S5.3 ChiSq test – LMOD1 (WH2 domain) (gnomAD v3.1.2) All TAAD cohort | |||
| Yes LMOD1 | No LMOD1 | Allele frequency | |
| TAAD (WH2) | 3 | 3499 | 0.00086 |
| gnomAD (WH2) | 3 | 152170 | 0.00002 |
Result: X2 (1, N = 155675) = 62.22, p<3.077e-15)
- Burden test (all TAAD cohort between TMBS + ABS1 domains)
A “burden test” was done with the “all TAAD cohort” with only the variants found between the TMBS and ABS1 domains of the LMOD1 gene using a public genomic database (gnomAD v3.1.2) in RStudio (v1.4.1717 [R version = R4.1.1]) with the following result: [X2 (1, N = 155704) = 3.821, p<0.05063)].
Data and the calculations
Total of 3 variants between the TMBS + ABS1 domains of the LMOD1 gene were found in 3 alleles (all were heterozygotes):
0/90 alleles in France
0/179 alleles in UK
0/88 alleles in Belgium-Ghent
2/1580 alleles in Belgium-Antwerp
1/1560 alleles in USA
0/2 alleles in Netherlands
Variants were classified as:
2 non-synonymous
1 nonsense
Total Probands: 1749.5 (1660 + 89.5) probands:
45 France
89.5 UK (this is an average)
44 Belgium-Ghent
790 Belgium-Antwerp
780 USA
1 Netherlands
The UK TAAD cohort allele final number (179) is an average from these variants:
Va47Leu – observed 1/182 alleles (91 probands)
Gly142Asp – observed 1/182 alleles (91 probands)
Thr185= – observed 1/182 alleles (91 probands)
Thr295Met – observed 46/166 alleles (83 probands) (TT = 5 / CT = 36 / CC= 42 – probands)
Prp544His – observed 1/182 alleles (91 probands)
Val595Ala – observed 1/180 alleles (90 probands)
Total alleles: 3499 alleles
International TAAD cohort 1660 * 2 = 3320 alleles
UK TAAD cohort 89.5 * 2 = 179 alleles
Genetic Database:
gnomAD (v3.1.2) all
Variant – Exclusion Criteria:
intron_variant
splice_region_variant
Variant – Inclusion Criteria:
Variant found between the TMBS and ABS1 domains (residues 1-100 of the protein)
MAF ≤0.00001315
Formula and command line for RStudio
>LMOD1_ChiSq_test <- matrix(c(3,3499,43,152159),ncol=2,byrow=TRUE); colnames(LMOD1_ChiSq_test) <- c(“Yes_LMOD1_variants”,”No_LMOD1_variants”); rownames(LMOD1_ChiSq_test) <- c(“TAAD_TMBS_ABS1″,”gnomAD_TMBS_ABS1”); LMOD1_ChiSq_test <- as.table(LMOD1_ChiSq_test); LMOD1_ChiSq_test; chisq <- chisq.test(LMOD1_ChiSq_test); chisq; expected <- as.array(margin.table(LMOD1_ChiSq_test,1)) %*% t(as.array(margin.table(LMOD1_ChiSq_test,2))) / margin.table(LMOD1_ChiSq_test); expected; chi <- sum((expected – as.array(LMOD1_ChiSq_test))^2/expected); chi; pchisq(chi,df=1,lower.tail = FALSE); ft <- fisher.test(LMOD1_ChiSq_test); ft; summary(LMOD1_ChiSq_test)
| Table S5.4 ChiSq test – LMOD1 (between TMBS and ABS1 domains) (gnomAD v3.1.2) All TAAD cohort | |||
| Yes LMOD1 | No LMOD1 | Allele frequency | |
| TAAD (TMBS_ABS1) | 3 | 3499 | 0.0009 |
| gnomAD (TMBS_ABS1) | 43 | 152159 | 0.0003 |
Result: X2 (1, N = 155704) = 3.821, p<0.05063)
- Burden test (all TAAD cohort in TMBS domain)
A “burden test” was done with the “all TAAD cohort” with only the variants found in the TMBS domain of the LMOD1 gene using a public genomic database (gnomAD v3.1.2) in RStudio (v1.4.1717 [R version = R4.1.1]) with the following result: [X2 (1, N = 155691) = 6.432, p<0.01121].
Data and the calculations
Total of 2 variants in the TMBS domain of the LMOD1 gene were found in 2 alleles (all were heterozygotes):
0/90 alleles in France
0/179 alleles in UK
0/88 alleles in Belgium-Ghent
1/1580 alleles in Belgium-Antwerp
1/1560 alleles in USA
0/2 alleles in Netherlands
Variants were classified as:
1 non-synonymous
1 nonsense
Total Probands: 1749.5 (1660 + 89.5) probands:
45 France
89.5 UK (this is an average)
44 Belgium-Ghent
790 Belgium-Antwerp
780 USA
1 Netherlands
The UK TAAD cohort allele final number (179) is an average from these variants:
Va47Leu – observed 1/182 alleles (91 probands)
Gly142Asp – observed 1/182 alleles (91 probands)
Thr185= – observed 1/182 alleles (91 probands)
Thr295Met – observed 46/166 alleles (83 probands) (TT = 5 / CT = 36 / CC= 42 – probands)
Prp544His – observed 1/182 alleles (91 probands)
Val595Ala – observed 1/180 alleles (90 probands)
Total alleles: 3499 alleles
International TAAD cohort 1660 * 2 = 3320 alleles
UK TAAD cohort 89.5 * 2 = 179 alleles
Genetic Database:
gnomAD (v3.1.2) all
Variant – Exclusion Criteria:
intron_variant
splice_region_variant
Variant – Inclusion Criteria:
Variant found in the TMBS domain (residues 1-41 of the protein)
MAF ≤0.00001315
Formula and command line for RStudio
>LMOD1_ChiSq_test <- matrix(c(2,3499,16,152174),ncol=2,byrow=TRUE); colnames(LMOD1_ChiSq_test) <- c(“Yes_LMOD1_variants”,”No_LMOD1_variants”); rownames(LMOD1_ChiSq_test) <- c(“TAAD_TMBS”,”gnomAD_TMBS”); LMOD1_ChiSq_test <- as.table(LMOD1_ChiSq_test); LMOD1_ChiSq_test; chisq <- chisq.test(LMOD1_ChiSq_test); chisq; expected <- as.array(margin.table(LMOD1_ChiSq_test,1)) %*% t(as.array(margin.table(LMOD1_ChiSq_test,2))) / margin.table(LMOD1_ChiSq_test); expected; chi <- sum((expected – as.array(LMOD1_ChiSq_test))^2/expected); chi; pchisq(chi,df=1,lower.tail = FALSE); ft <- fisher.test(LMOD1_ChiSq_test); ft; summary(LMOD1_ChiSq_test)
| Table S5.5 ChiSq test – LMOD1 (TMBS) (gnomAD v3.1.2) All TAAD cohort | |||
| Yes LMOD1 | No LMOD1 | Allele frequency | |
| TAAD (TMBS) | 2 | 3499 | 0.0006 |
| gnomAD (TMBS) | 16 | 152174 | 0.0001 |
Result: X2 (1, N = 155691) = 6.432, p<0.01121)
Supplement 6
Copy Number Variation (CNV)
CNV was assessed in the UK TAAD family M441 by exome depth software and rare and previously unreported deletions, or duplications were considered candidates. There was no evidence of co-segregation in the 3 individuals (IV:2, IV:5 and IV:16) (figure 1A) of any structural variation with the phenotype.
CNV was not assessed in the Dutch TAAD family Netherlands01.