
Abstract
Brugada syndrome is a genetic condition that predisposes to ventricular tachycardia. It is caused by loss of function heterozygous mutations of the SCN5A gene in 20% of patients with the condition. There is only a single report of Brugada syndrome due to a large deletion involving the whole of SCN5A in the literature. We describe a further two families with identical chromosome 3p22.2 deletions including the SCN5A and SCN10A genes only.
Affected family members had clinical features of Brugada syndrome, conduction disease, ventricular arrhythmias and sudden death. The deletion was initially identified using MLPA and array CGH. Medium coverage whole genome sequencing was used to define the breakpoints of the deletion, confirming involvement of both SCN5A and SCN10A, and that the deletions were identical in the two families. A facile PCR-based assay was developed for cascade testing of additional family members.
This report confirms that heterozygous whole gene deletions involving SCN5A cause Brugada syndrome and overlapping phenotypes. Deletion of SCN10A may also be contributing to the cardiac phenotype. This study also highlights the utility of medium coverage whole genome sequencing for defining deletion breakpoints in a diagnostic setting.
Author Information
Brugada syndrome caused by a 3p22.2 deletion including SCN5A and SCN10A defined using medium coverage whole exome sequencing
Rosalyn Jewell1*, Susan Clasper2, Stephen P. Page3, Christopher M. Watson4,5, Oonagh Claber6, Christopher Hayes7, Kate Sergeant2, Andrea Coates5, Laura A. Crinnion4,5, Sarah Hewitt5, Matthieu J. Miossec8,9, Mauro Santibanez-Koref8, Kathryn Ashcroft1, David T. Bonthron1,4, Bernard Keavney10, Paul Brennan6, Jennifer Thomson1
- Yorkshire Regional Genetics Service, Leeds Teaching Hospitals NHS Trust, Leeds, LS7 4SA, UK
- Genetics Laboratories, Oxford University Hospitals NHS Trust, Oxford, OX3 7LE, UK
- Department of Cardiology, Leeds Teaching Hospitals NHS Trust, Leeds, LS1 3EX, UK
- School of Medicine, University of Leeds, St James’s University Hospital, Leeds, LS9 7TF, UK
- Leeds Genetics Laboratories, Leeds Teaching Hospitals NHS Trust, Leeds, LS9 7TF, UK
- Northern Genetics Service, Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, NE1 3BZ, UK
- Department of Cardiology, York Teaching Hospital NHS Foundation Trust, York, YO31 8HE, UK
- Institute of Human Genetics, Newcastle University, Newcastle upon Tyne NE1 3BZ, UK
- Center for Bioinformatics and Integrative Biology, Faculty of Biological Sciences, Universidad Andrés Bello, Santiago, Chile
- Institute of Cardiovascular Sciences, University of Manchester, Manchester, M13 9NT, UK
Corresponding author:
*Dr Rosalyn Jewell
Yorkshire Regional Genetics Service,
3rd Floor, Chapel Allerton Hospital,
Chapel Town Road,
Leeds, LS7 4SA
Email: rosalyn.jewell@nhs.net
Tel: 01133924415
Fax: 01133924434
Journal subject terms:
Arrhythmias, genetics
Key words:
Brugada syndrome, 3p22.2, deletion, SCN5A
Date submitted- 17th August 2018
Date accepted- 12th October 2018
Background
Brugada syndrome (OMIM: 601144) was first reported in 1992 and is characterised by coved ST segment elevation in anterior precordial leads and tendency to ventricular tachycardia (Chockalingam and Wilde, 2012, Brugada and Brugada, 1992). This autosomal dominant condition with variable penetrance can be caused by mutations in genes that encode sodium channels. Mutations in the SCN5A gene are identified in approximately 20% of patients with the condition (Kapplinger et al., 2010, Meregalli et al., 2009, Ruan et al., 2009, Jeevaratnam et al., 2016). Loss of function SCN5A mutations, leading to haploinsufficiency of the sodium voltage-gated channel alpha subunit 5 (NaV1.5), are thought to cause depolarization abnormalities and altered conduction or repolarization abnormalities leading to a phase II re-entry tendency. The mechanisms leading to arrhythmias in Brugada syndrome are multifactorial with older age and male sex being associated with an increased tendency to arrhythmias (Jeevaratnam et al., 2016).
There are several case reports of intragenic deletions or rearrangements in the SCN5A gene being associated with Brugada syndrome (Eastaugh et al., 2011, Zumhagen et al., 2013, Koopmann et al., 2007, Garcia-Molina et al., 2013, Allegue et al., 2015). Recently, there has been a single report of a chromosome 3p22.2 deletion involving SCN5A and an additional seven genes, identified using sequencing technology and confirmed with copy number analysis using SNP-array (Trujillo-Quintero et al., 2018).
Here we report two families affected with Brugada syndrome caused by a 3p22.2 deletion including the SCN5A and SCN10A genes. The deletion was originally identified using multiplex ligation-dependent probe amplification (MLPA) analysis of SCN5A. Array comparative genomic hybridization (aCGH) subsequently confirmed that the deleted region included SCN10A. To map the deletion breakpoints at nucleotide resolution and enable development of a facile PCR-based assay for cascade testing of additional family members, medium coverage whole genome sequencing was performed. This confirmed that the deletions are identical in both families, suggesting they are related, although this has not been confirmed on pedigree analysis.
Methods
Probands were identified in genetics clinics held in the North of England as part of NHS Regional Clinical Genetics Services. Both families provided informed consent for the analyses described and for publication of results. DNA was extracted from peripheral blood lymphocytes of recruited family members using a standard salting out method.
SCN5A gene mutation analysis
Mutation screening was performed on the proband from family 1 by Sanger fluorescent sequencing exon/intron boundaries and coding nucleotides of SCN5A exons 2-28 (NM_198056.2) using standard PCR primer sequences and thermocycling conditions.
Dosage analysis to identify a deletion or duplication of one or more exons was performed using a MLPA assay (kit P108-B2) (MRC-Holland, Netherlands). Analysis was performed using Coffalyser software.
Array comparative genomic hybridization (aCGH) analysis
Targeted aCGH analysis was performed using a BlueGnome ISCA 8 × 60k OligoArray (v2.0) (BlueGnome Ltd., Cambridge, UK). Manufacturer’s protocols were followed throughout. The reference control sample comprised a mixed pool of five healthy sex matched individuals (Promega U.K., Southampton, UK). Data analysis was performed using BlueFuse Multi software (v4.1) (BlueGnome Ltd., Cambridge, UK) and the Decipher database was interrogated to aid variant interpretation (https://decipher.sanger.ac.uk) (Firth et al., 2009).
Medium coverage whole genome sequencing (WGS)
To determine the precise deletion breakpoints medium coverage WGS was performed. Approximately 1 μg of genomic DNA was sheared using a CovarisS2 (Covaris, Inc., Woburn, MA, USA) before an Illumina-compatible sequencing library was generated using NEBNext® UltraTM reagents (New England Biolabs, Ipswich, MA, USA). A size selection which aimed to generate insert fragments of between ~300-400bp and 6 rounds of thermocycling were performed. All other steps were followed as per the manufacturer’s protocol. The final library was checked on an Agilent Bioanalyser (Agilent Technologies, Stockport, UK) prior to sequencing on a single lane of a HiSeq2500 Rapid Mode flow cell configured to generate paired end 101bp reads (Illumina, Inc., San Diego, CA, USA). Raw sequence data was converted to fastq.gz format using bcl2fastq v.2.17.14. The per-sample sequence reads were aligned to an indexed human reference genome (hg19) using bwa v.0.7.13 (http://bio-bwa.sourceforge.net) (Li and Durbin, 2009). Duplicate reads were marked, and sorted by chromosome coordinate, using Picard v.2.4.1 (https://broadinstitute.github.io/picard/). Reads with a mapping quality ≥1 were extracted from the approximate location of the aCGH defined deleted region (chr3:38551000-38857000) using samtools v.0.1.18 (http://www.htslib.org) (Li et al., 2009). These data were interrogated to identify read pairs spanning the deletion breakpoint. The mapped reads in the duplicate removed coordinate-sorted BAM file were visualised using the Integrative Genome Browser (http://software.broadinstitute.org/software/igv/) (Thorvaldsdottir et al., 2013). Breakpoint spanning reads were identified following interpretation of the alignment CIGAR string. BLAT was then used to determine precise the genomic coordinates of the 5′ and 3′ fragments (http:// genome.ucsc.edu) (Kent, 2002).
Molecular confirmation of the deletion breakpoint
A PCR amplicon was designed to amplify across the deletion breakpoint. The primer sequences used to amplify the deleted allele were dTGTAAAACGACGGCCAGTACTCAGTGATGGTGGTAATGTT (common forward) and dCAGGAAACAGCTATGACCAGTAAGGGTCTGGGAAGCTG (reverse deletion) which generated a 594-bp PCR product. A second reverse primer dCAGGAAACAGCTATGACCGTGGCAGCACAAACCCTG (reverse normal) was designed to work in combination with the common forward primer to amplify the normal allele. This reaction generated a smaller 436-bp PCR product. All primers contained universal tags (underlined) to enable Sanger sequencing using our routine laboratory workflow. The multiplex PCR reaction comprised 0.5 μl of genomic DNA (406 ng/μl), 11 μl of MegaMix PCR reagent (Microzone Ltd., Haywards Heath, UK), 1 μl of 10 μM common forward primer, 2 μl of 10 μM reverse deletion primer, and 0.5 μl of 10 μM reverse normal primer. Thermocycling conditions consisted of 5 minutes at 94 ΟC then 30 cycles of 94 ΟC for 30 seconds, 60 ΟC for 1 minute, and 72 ΟC for 45 seconds before a final extension step at 72 ΟC for 5 minutes. PCR products were resolved on a 2% tris-borate EDTA agarose gel. Sanger sequencing was performed on an ABI3730 to confirm the identity of both PCR amplicons; manufacturer’s protocols were followed throughout (Life Technologies Ltd., Paisley, UK). Sequence chromatograms were analysed using Chromas Lite v.2.1.1 (http://technelysium.com.au/wp/chromas/).
BED files containing the genomic start and stop coordinates of deleted sequence data corresponding to specific assays were generated and displayed using the UCSC genome browser (http://genome.ucsc.edu) (Kent et al., 2002).
Results
Family 1 clinical features
The male proband presented at the age of 36 years with central chest pain preceded by two episodes of syncope. An electrocardiogram (ECG) showed 3mm of J point elevation in V1 with coved ST segments and an inverted T wave consistent with a type I ECG pattern. ECGs with high ventricular leads confirmed this appearance in V1 and 2mm of J point elevation appearance of flat ST segments and an inverted wave in V2. There was no fractionation.
The patient had no other medical history and no history of developmental delay, intellectual disability or behavioural issues. He had no dysmorphic features. Following discussion of risk of sudden cardiac death, the patient declined an implantable cardiac defibrillator (ICD).
The pedigree for the family is presented in Figure 1. Familial screening revealed the proband’s mother also had a clinical diagnosis of Brugada syndrome. The proband’s maternal grandfather died of Parkinson’s disease and grandmother was reported to have heart problems. The proband has two brothers, one of whom had a normal ajmaline test and the other declined to be reviewed.
Genetic testing results for family 1
The proband proceeded with genetic testing for Brugada syndrome, which included Sanger sequencing and MLPA analysis of SCN5A. A heterozygous whole gene deletion was identified comprising 31 deleted probes spanning the SCN5A locus (Supplementary Figure 1). This defined a minimally deleted region of approximately 101kb.
In order to confirm this finding, and determine the maximal extent of the deleted region, aCGH was performed identifying a heterozygous deletion at 3p22.2 and defined deleted probes whose minimal and maximal intervals were 157 kb and 296 kb respectively (genomic coordinates 38603213-38759936 and 38560654-38856305).
Although the resolution of this assay was too low to confirm an SCN5A whole gene deletion it did confirm a deletion of at least exons 1-22 (NM_198056.2). Furthermore, the minimally deleted aCGH region encompassed exons 21-27 of the adjacent, proximally located, SCN10A gene (NM_006514.3).
In order to define the deletion breakpoints further, medium coverage (mean 8.5×) whole genome sequencing was performed. Summary sequencing metrics generated using our standard bioinformatics pipeline are recorded in Supplementary Table 1. Sequence reads with a MAQ score ≥1 and whose start position mapped within the genomic interval defined by the non-deleted aCGH probes were extracted from the duplicate removed coordinate-sorted BAM file; this yielded 12,600 reads. Of these, 5 read pairs had insert sizes ranging between 282,491 bp and 282,953 bp which were consistent with the extent of the aCGH defined deleted interval. The mapped positions of the proximally aligned reads were located within a 210bp interval (Table 1). Two read pairs contained clipped sequences which, following BLAT analysis, were determined to span the deletion breakpoint (Supplementary Table 2). The substantial decrease in the read density across the site of the deleted region is shown in Supplementary figure 2.
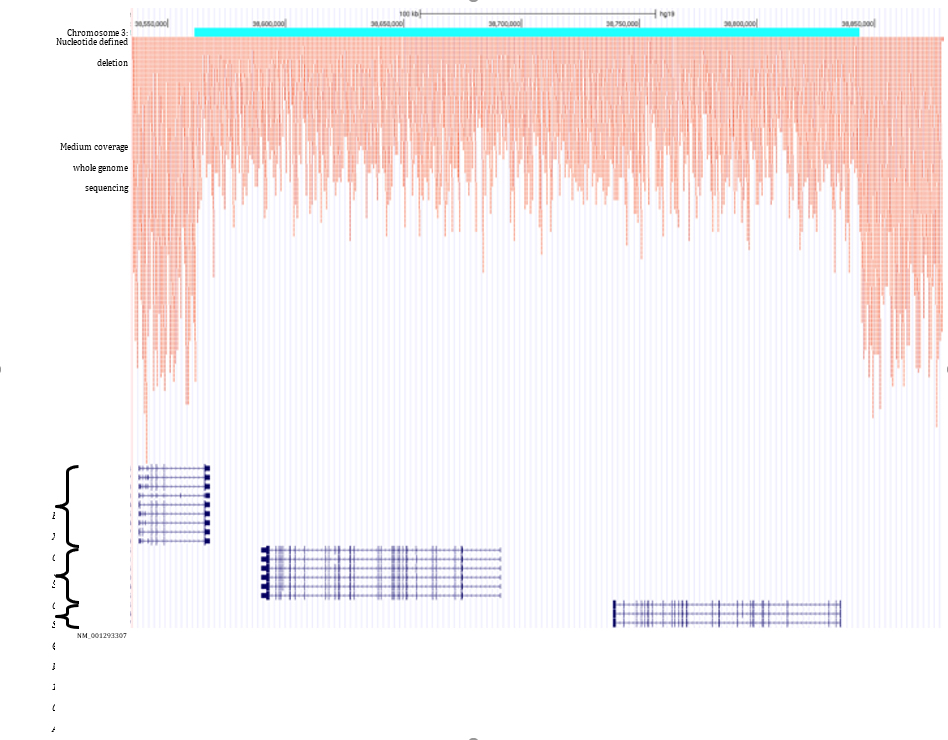
The 5′ deletion breakpoint is located within the EXOG gene and the 3′ breakpoint lies beyond the SCN10A start site (Figure 2). When viewed in the context of the RepeatMasker tract the 5′ deletion site intersects the end of a Charlie 16a DNA repeat and the 3′ breakpoint intersects an L2 LINE element.
To validate the deletion, a breakpoint spanning PCR assay was designed. Sanger sequencing of the amplicon confirmed a clean deletion breakpoint with no inserted nucleotides (Figure 3). A multiplex PCR was subsequently optimised incorporating a normal allele-specific reverse primer to work in conjunction with the deletion-specific reverse primer. This provided a single assay to determine the zygosity and detect the mutation in additional family members (Figure 4).
Family 2
Interrogation of the Decipher database identified a second family with a similar deletion (Firth et al., 2009). The male proband presented in his 5th decade following two syncopal episodes secondary to ventricular fibrillation. He was diagnosed with idiopathic VF and found to have ST elevation and right bundle branch block on his ECG. He was treated with beta-blockers and an ICD.
Review of the pedigree (Figure 1), revealed that the proband’s sister also suffered with syncopal episodes due to spontaneous VF and had an ICD. A further male sibling also had an ICD and another male sibling died at the age of 49, secondary to left ventricular failure and ischaemic heart disease.
More extensive screening of children and grandchildren revealed a number of affected family members with abnormal ECG findings (Figure 1and Figure 5). Affected family members were treated with ICDs and beta-blockers.
Genetic testing results for family 2
DNA samples from family 2 were initially sent for SCN5A sequencing in 2000 and these were normal.
Subsequently, in 2014, a child was born with multiple congenital anomalies (indicated on the pedigree in Figure 1). aCGH was performed which identified a deletion at chromosome 3p22.3. This was not related to the child’s phenotype, but included the SCN5A gene and was clearly of relevance to the features of Brugada syndrome in the extended family.
Genetic testing was offered to family members affected with cardiac features and those at risk of having Brugada syndrome. This identified a number of family members with the 3p22.3 deletion, some of whom had no clinical symptoms or features of Brugada syndrome on 12-lead ECG (Figure 1).
A parallel effort to identify genetic variants in 5 individuals from family 2 using whole-exome sequencing also lead to the detection of the deletion spanning SCN5A. Notably, this deletion was also found to encompass SCN10A (Supplementary Figure 3).
Application of the breakpoint spanning PCR developed for family 1 to samples from members of family 2 demonstrates that the deletion is identical in the two families (Figure 4).
Discussion
We have described two families from Northern England with an identical chromosome 3p22.2 deletion resulting in loss of the SCN5A and SCN10A genes.
The phenotypes observed in these two families are a combination of Brugada pattern ECGs, conduction disease, ventricular arrhythmias and sudden death. Overlap phenotypes are being increasingly recognised in families with SCN5A gene mutations, which is important to appreciate when screening at risk relatives. Risk stratification is also very difficult as the conventional risk markers in Brugada syndrome (syncope and spontaneous Type 1 Brugada pattern) may be absent in individuals with marked conduction disease. The coexistence of conduction disease and Brugada type ECG changes can also make ajmaline provocation hazardous so the risks and benefits of testing in this situation need to be carefully considered.
Heterozygous deletions of SCN5A, which has a haploinsuffiency score of 15.55% would be predicted to have a similar functional effect to loss of function mutations of SCN5A that have been previously associated with Brugada syndrome (Huang et al., 2010).
The deletion described here also involves loss of the SCN10A gene. This encodes the sodium voltage-gated channel alpha subunit 10 (NaV1.8). This subunit is highly expressed in nociceptive sensory neurons of dorsal root ganglia and cranial sensory ganglia with gain of function mutations in SCN10A being associated with an episodic pain disorder (Faber et al., 2012, Akopian et al., 1996). SCN10A is also expressed in cardiac neurones (Verkerk et al., 2012), myocardium (Yang et al., 2012, Chambers et al., 2010) and the cardiac conduction system (Sotoodehnia et al., 2010, Pallante et al., 2010). NaV1.8 and NaV1.5, encoded by SCN5A, associate with each other in the plasma membrane (Hu et al., 2014) and are likely to be subject to common regulatory mechanisms, for example transcriptional control by TBX3 or TBX5 (van den Boogaard et al., 2014).
Polymorphisms in the SCN5A/SCN10A region have been reported as associated with haplotypes leading to PR- and QRS- prolongation (van den Boogaard et al., 2014, Pfeufer et al., 2009, Sotoodehnia et al., 2010), with a genome wide association study showing a significant association between polymorphisms at the SCN10A locus and Brugada syndrome (Bezzina et al., 2013). More recently, variants in SCN10A have been identified in patients with Brugada syndrome. It has been suggested that such variants lead to loss of NaV1.8 function or alternatively modify the function of NaV1.5. Further functional studies are however required and the clinical significance of such SCN10A variants remains controversial (Fukuyama et al., 2015, Behr et al., 2015, Hu et al., 2014, Fernandez-Falgueras et al., 2017, Gourraud et al., 2016).
We propose that the heterozygous deletion of SCN5A is contributing to the Brugada phenotype in the families described and may be modified by syntenic deletion of SCN10A. The clinical features in some family members are, however, consistent with classical Brugada syndrome, suggesting that the phenotype has been more determined by haploinsufficiency of SCN5A than SCN10A.
Recently, there has been a report of a family with a larger deletion of 3p22.2 including SCN5A (Trujillo-Quintero et al., 2018). This was initially identified using next generation sequencing of a panel of genes and then confirmed using SNP-array in contrast to the approach described in this report. The deletion was larger (643Kb) than the one described here (282Kb) and involved eight genes, including SCN5A and SCN10A. The family described had Brugada syndrome, confirming the current report that whole gene deletions of SCN5A cause Brugada syndrome and should be regarded as pathogenic (Trujillo-Quintero et al., 2018).
WGS is being utilised in the 100,000 Genomes Project in England and other studies to identify point mutations at read depths of around 30x. This report describes a use for WGS to identify breakpoints at nucleotide resolution with 8x coverage. This approach has been previously utilised for identification of mutations for single gene disorders (Watson et al., 2016). This report represents a further cost-effective and useful practical application for WGS in diagnostic testing.
WGS has demonstrated the deletions are identical in the two families. These families were seen in genetics centres in the north of England. In view of the lack of reports of this nature in the literature and the geographical proximity of the families, it is likely they are related. We have not, however, been able to demonstrate this via pedigree analysis. WGS did demonstrate regions of low complexity repeats around the deletion, which may explain the mechanism by which the deletion occurred via meiotic non-allelic homologous recombination (Gu et al., 2008, Weckselblatt and Rudd, 2015) Despite this, the likelihood of an identical, previously unreported deletion, occurring spontaneously in two unrelated families is low.
With use of aCGH, sequencing of larger panels of genes, exome and whole genome analysis, further deletions involving SCN5A will be identified. Diagnostic testing for Brugada syndrome needs to identify sequence variants and copy number changes in a cost effective manner. This is likely to be achieved using advances in sequencing technology and novel approaches, such as those described in this report.
Acknowledgements
We are very grateful to the families involved in this study.
Funding sources
None.
Disclosures
None.
Table 1. Five discordant read pairs whose fragment insert sizes fall within the aCGH defined deleted interval
| Read pair index | Read pair ID | Insert size (bp) | Read 1 | Read 2 | ||||||
| Position | Strand | MAQ | CIGAR | Position | Strand | MAQ | CIGAR | |||
| 1 | SN7001297:416:HVWMJBCXX:2:1204:15916:75216 | 282,491 | 38,843,561 | – | 60 | 49S52M | 38,561,122 | + | 60 | 101M |
| 2 | SN7001297:416:HVWMJBCXX:2:1208:5151:59669 | 282,617 | 38,560,976 | + | 60 | 100M | 38,843,561 | – | 60 | 68H32M |
| 3 | SN7001297:416:HVWMJBCXX:2:2201:2889:93193 | 282,634 | 38,561,099 | + | 60 | 101M | 38,843,633 | – | 60 | 100M |
| 4 | SN7001297:416:HVWMJBCXX:2:2212:7842:84077 | 282,739 | 38,561,129 | + | 60 | 101M | 38,843,768 | – | 60 | 100M |
| 5 | SN7001297:416:HVWMJBCXX:2:2116:19021:33945 | 282,953 | 38,560,710 | + | 60 | 99M | 38,843,562 | – | 60 | 101M |
MAQ: Mapping quality score; M: Matched nucleotides; S: soft-clipped nucleotides; H: hard-clipped nucleotides. Genomic positions are for chromosome 3 and are reported using human genome build hg19.
Figure legends
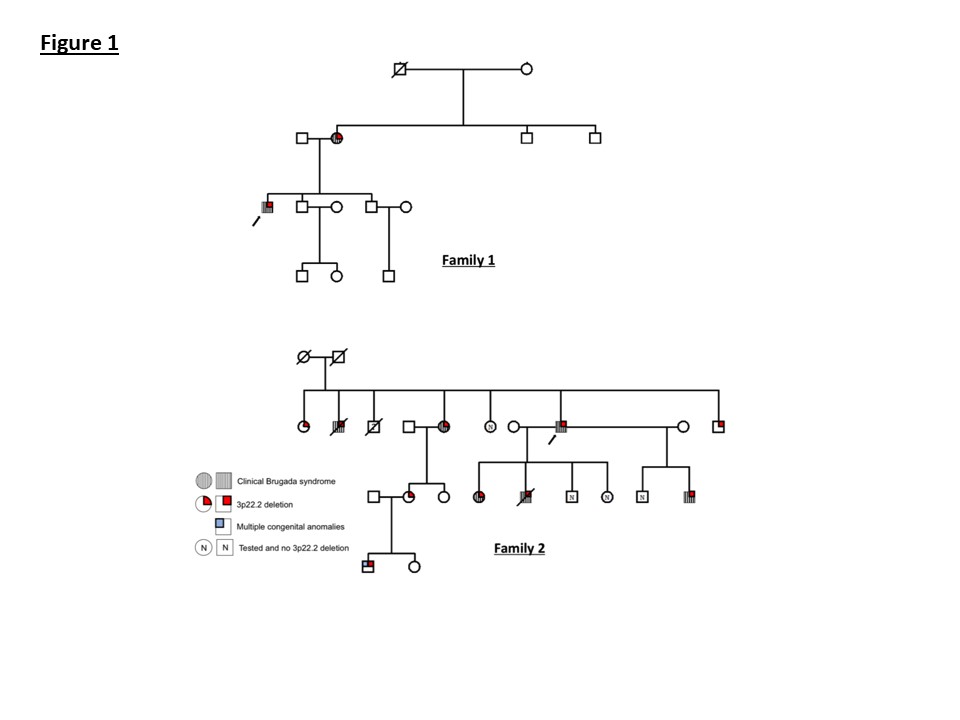
Figure 1: Pedigrees for families 1 and 2. The proband in each family is indicated by an arrow.
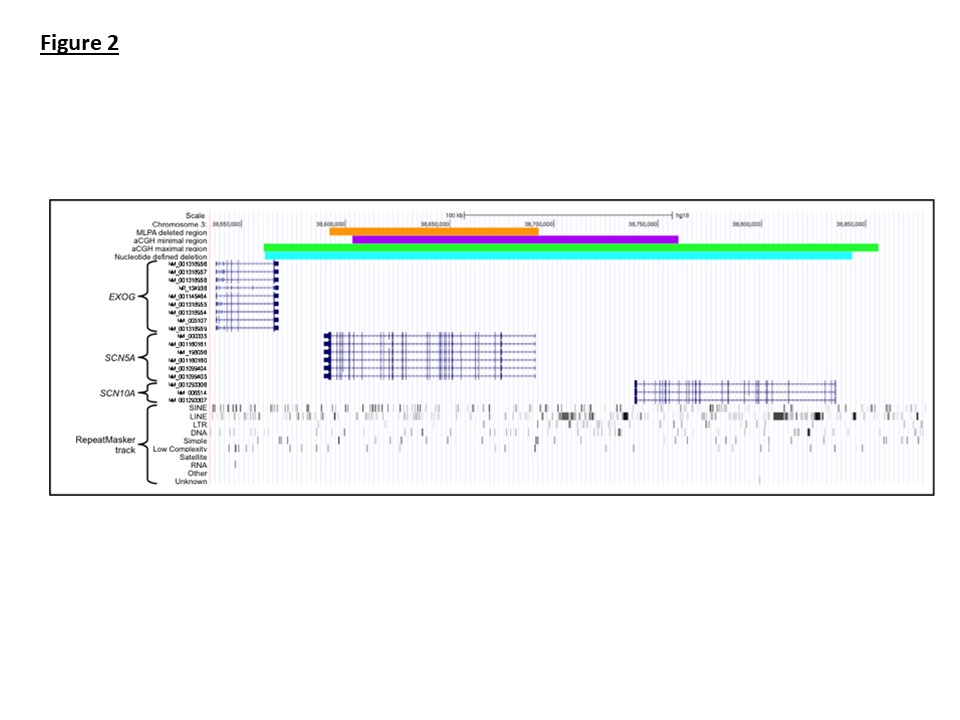

Figure 2: Visualization of the 3p22.2 region using the UCSC genome browser. The orange track displays the extent of MLPA defined deletion identified in the family 1 proband. The purple and green tracks show the minimum and maximum extents of the deletion when assessed by aCGH. The alignment start sites of whole genome sequencing reads are displayed in red. The cyan track displays the genomic coordinates of the nucleotide resolved deletion. Relative positions of the EXOG, SCN5A and SCN10A genes within the region are displayed using the RefSeq track.
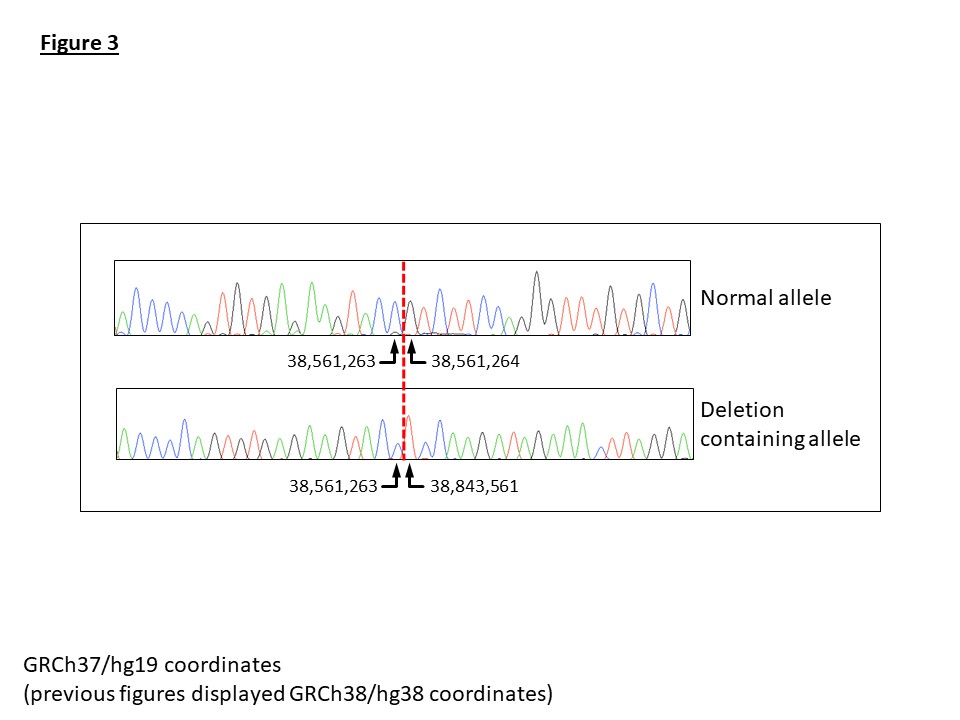
Figure 3: Sanger sequencing results for the normal and deletion containing amplicons. The dashed vertical red line indicated the location of the deletion breakpoint. Genomic coordinates are displayed for genome build hg19.
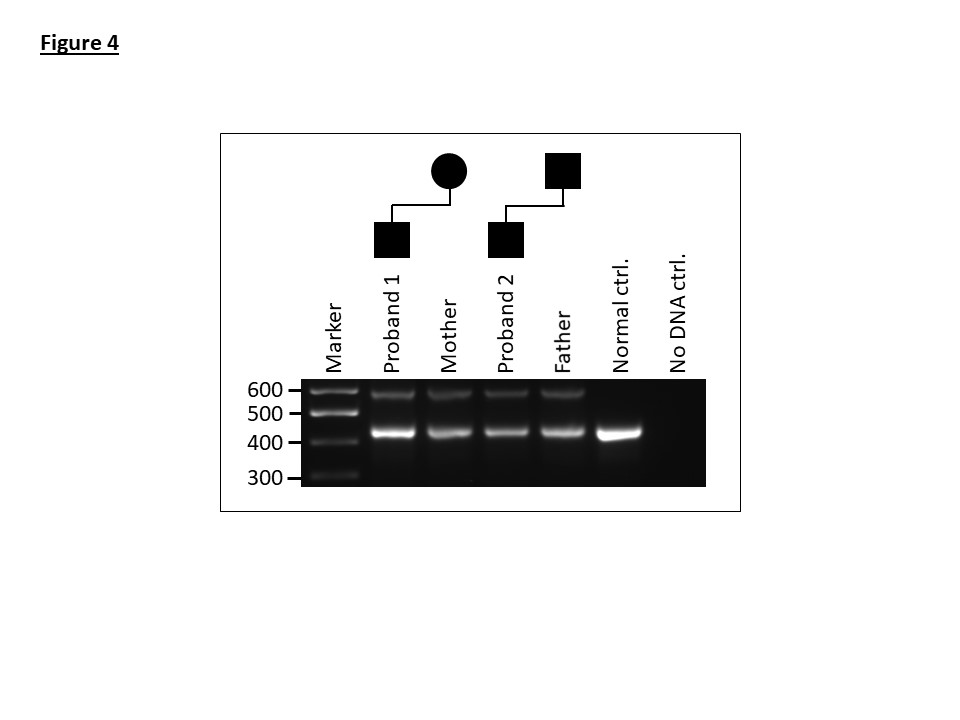
Figure 4: A multiplex PCR assay showing amplification products for the normal (463-bp) and deletion containing alleles (594-bp) for affected members of family 1 and 2.
Figure 5: ECGs.
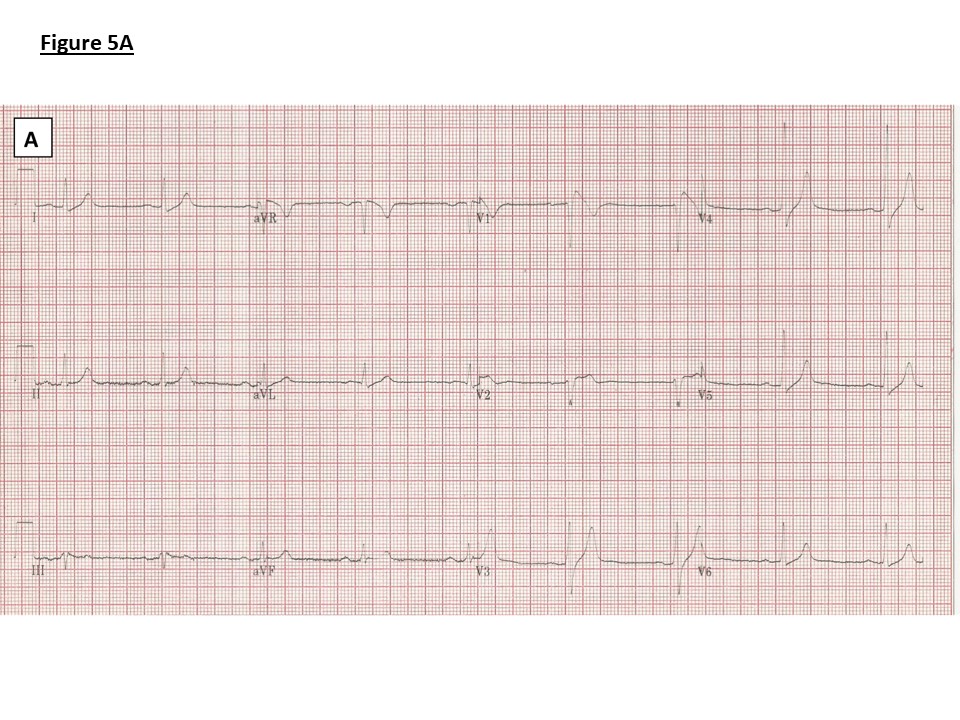
A) Family 1: ECG showing borderline PR prolongation (198ms), QRS broadening (114ms) and J point elevation, coved ST segment and T wave inversion in V1 (Type 1 Brugada pattern)
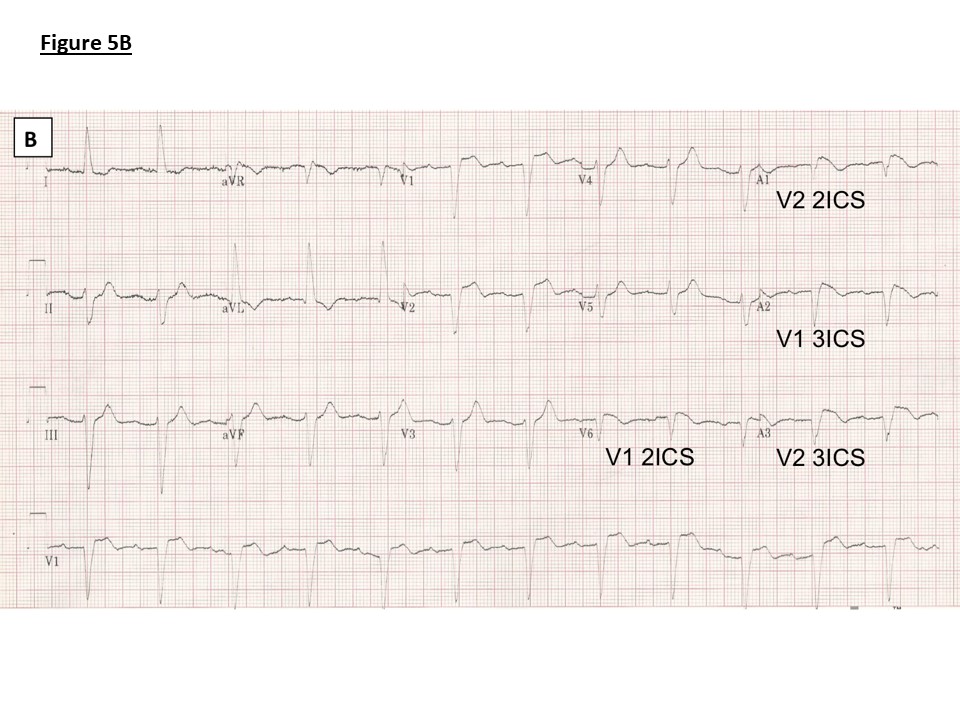
B) Family 1: ECG with high V1 and V2 leads as marked following ajmaline provocation showing PR prolongation (240ms), left axis deviation, left bundle branch block (QRS duration 146ms) and J point elevation, coved ST segment and T wave inversion in V1 (Type 1 Brugada pattern) V1 and V2 in 2nd and 3rd intercostal spaces
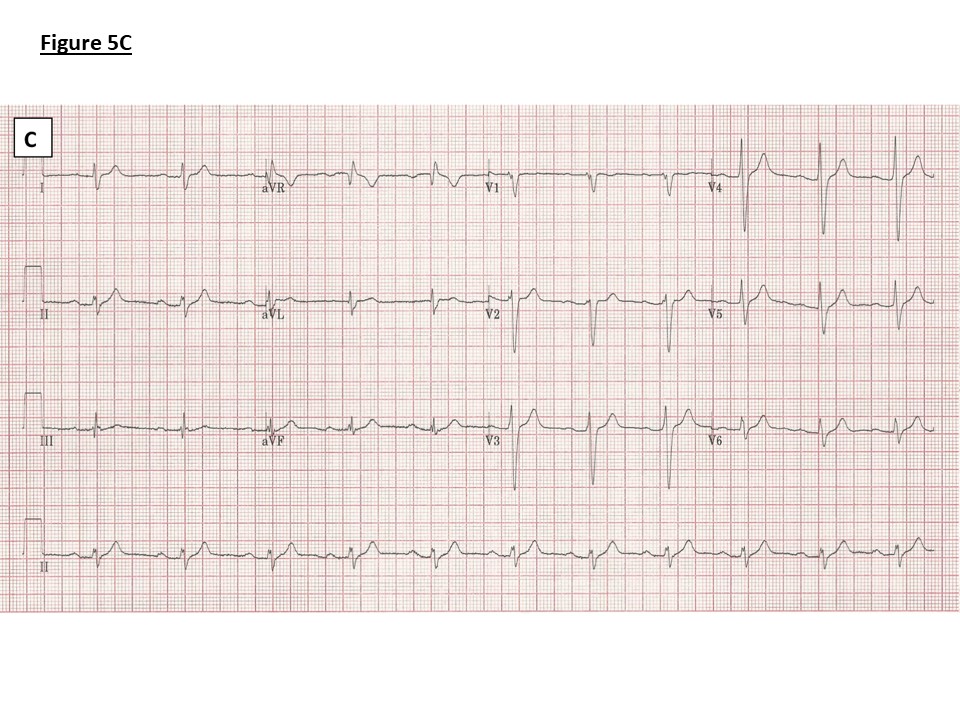
C) Family 2: ECG showing PR prolongation (236ms), right axis deviation and an intraventricular conduction delay (QRS duration 136ms). No J point elevation.
SUPPLEMENTARY MATERIAL
Supplementary Table 1. Sequencing and alignment metrics
| Total raw read pairs | Adaptor trimmed reads | Duplicate rate (%) | Median insert size (bp) | Mapped reads | Unmapped reads | |
| Read 1 (%) | Read 2 (%) | |||||
| 126,929,176 | 1,598,681 (1.3) | 1,605,808 (1.3) | 1.41 | 358 | 251,580,216 | 2,158,138 |
Supplementary Table 2. Characteristics of clipped breakpoint-spanning reads.
| Read pair index | Read pair ID | Sequence | Aln
(nts) |
Cl
(nts) |
Aln
start |
Aln
End |
Aln
str |
Cl
start |
Cl
end |
Cl str |
| 1 | SN7001297:416:HVWMJBCXX:2:1204:15916:75216 | GGAGGAAGATTTGAGGAAGCATAAAGGAAGACTCCTAAGTTTCTATCTTGGAGGTACTTCTCACACTGGGGTACCTGTTCCAGGCCAAAGGGTAGCAGCCT | 52 | 49 | 38,843,612 | 38,843,561 | – | 38,561,263 | 38,561,215 | – |
| 2 | SN7001297:416:HVWMJBCXX:2:1208:5151:59669 | ATAAAGGAAGACTCCTAAGTTTCTATCTTGGAGGTACTTCTCACACTGGGGTACCTGTTCCAGGCCAAAGGGTAGCAGCCTACTAAGTCACAGTAAAACA | 32 | 68 | 38,843,592 | 38,843,561 | – | 38,561,263 | 38,561,196 | – |
Aln: Aligned; Cl: Clipped; str: strand. Coordinate start and stop sites are for chromosome 3 and are reported for human genome build hg19
Supplementary Figure 1: MLPA analysis of SCN5A. A heterozygous whole gene deletion was identified comprising 31 deleted probes spanning the SCN5A locus.
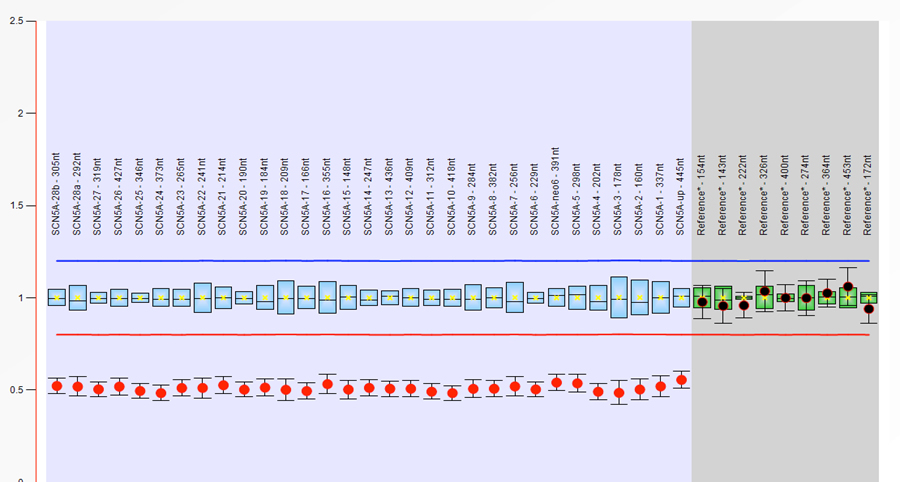
Supplementary Figure 2: Read density of medium coverage whole genome sequencing across the site of the deleted region. A substantial decrease in read density is seen.
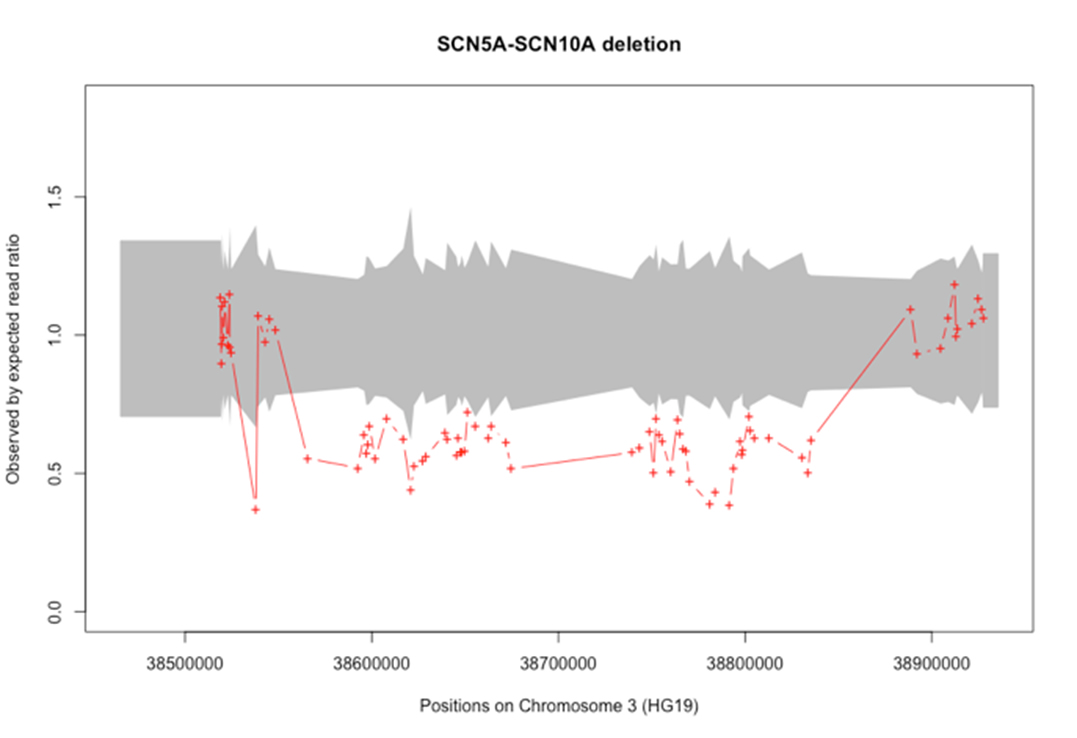
Supplementary Figure 3: Deletion identified in family 2 using the ExomeDepth R package.
References
AKOPIAN, A. N., SIVILOTTI, L. & WOOD, J. N. 1996. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature, 379, 257-62.
ALLEGUE, C., COLL, M., MATES, J., CAMPUZANO, O., IGLESIAS, A., SOBRINO, B., BRION, M., AMIGO, J., CARRACEDO, A., BRUGADA, P., BRUGADA, J. & BRUGADA, R. 2015. Genetic Analysis of Arrhythmogenic Diseases in the Era of NGS: The Complexity of Clinical Decision-Making in Brugada Syndrome. PLoS One, 10, e0133037.
BEHR, E. R., SAVIO-GALIMBERTI, E., BARC, J., HOLST, A. G., PETROPOULOU, E., PRINS, B. P., JABBARI, J., TORCHIO, M., BERTHET, M., MIZUSAWA, Y., YANG, T., NANNENBERG, E. A., DAGRADI, F., WEEKE, P., BASTIAENAN, R., ACKERMAN, M. J., HAUNSO, S., LEENHARDT, A., KAAB, S., PROBST, V., REDON, R., SHARMA, S., WILDE, A., TFELT-HANSEN, J., SCHWARTZ, P., RODEN, D. M., BEZZINA, C. R., OLESEN, M., DARBAR, D., GUICHENEY, P., CROTTI, L., CONSORTIUM, U. K. & JAMSHIDI, Y. 2015. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res, 106, 520-9.
BEZZINA, C. R., BARC, J., MIZUSAWA, Y., REMME, C. A., GOURRAUD, J. B., SIMONET, F., VERKERK, A. O., SCHWARTZ, P. J., CROTTI, L., DAGRADI, F., GUICHENEY, P., FRESSART, V., LEENHARDT, A., ANTZELEVITCH, C., BARTKOWIAK, S., BORGGREFE, M., SCHIMPF, R., SCHULZE-BAHR, E., ZUMHAGEN, S., BEHR, E. R., BASTIAENEN, R., TFELT-HANSEN, J., OLESEN, M. S., KAAB, S., BECKMANN, B. M., WEEKE, P., WATANABE, H., ENDO, N., MINAMINO, T., HORIE, M., OHNO, S., HASEGAWA, K., MAKITA, N., NOGAMI, A., SHIMIZU, W., AIBA, T., FROGUEL, P., BALKAU, B., LANTIERI, O., TORCHIO, M., WIESE, C., WEBER, D., WOLSWINKEL, R., CORONEL, R., BOUKENS, B. J., BEZIEAU, S., CHARPENTIER, E., CHATEL, S., DESPRES, A., GROS, F., KYNDT, F., LECOINTE, S., LINDENBAUM, P., PORTERO, V., VIOLLEAU, J., GESSLER, M., TAN, H. L., RODEN, D. M., CHRISTOFFELS, V. M., LE MAREC, H., WILDE, A. A., PROBST, V., SCHOTT, J. J., DINA, C. & REDON, R. 2013. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet, 45, 1044-9.
BRUGADA, P. & BRUGADA, J. 1992. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol, 20, 1391-6.
CHAMBERS, J. C., ZHAO, J., TERRACCIANO, C. M., BEZZINA, C. R., ZHANG, W., KABA, R., NAVARATNARAJAH, M., LOTLIKAR, A., SEHMI, J. S., KOONER, M. K., DENG, G., SIEDLECKA, U., PARASRAMKA, S., EL-HAMAMSY, I., WASS, M. N., DEKKER, L. R., DE JONG, J. S., STERNBERG, M. J., MCKENNA, W., SEVERS, N. J., DE SILVA, R., WILDE, A. A., ANAND, P., YACOUB, M., SCOTT, J., ELLIOTT, P., WOOD, J. N. & KOONER, J. S. 2010. Genetic variation in SCN10A influences cardiac conduction. Nat Genet, 42, 149-52.
CHOCKALINGAM, P. & WILDE, A. 2012. The multifaceted cardiac sodium channel and its clinical implications. Heart, 98, 1318-24.
EASTAUGH, L. J., JAMES, P. A., PHELAN, D. G. & DAVIS, A. M. 2011. Brugada syndrome caused by a large deletion in SCN5A only detected by multiplex ligation-dependent probe amplification. J Cardiovasc Electrophysiol, 22, 1073-6.
FABER, C. G., LAURIA, G., MERKIES, I. S., CHENG, X., HAN, C., AHN, H. S., PERSSON, A. K., HOEIJMAKERS, J. G., GERRITS, M. M., PIERRO, T., LOMBARDI, R., KAPETIS, D., DIB-HAJJ, S. D. & WAXMAN, S. G. 2012. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A, 109, 19444-9.
FERNANDEZ-FALGUERAS, A., SARQUELLA-BRUGADA, G., BRUGADA, J., BRUGADA, R. & CAMPUZANO, O. 2017. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology (Basel), 6.
FIRTH, H. V., RICHARDS, S. M., BEVAN, A. P., CLAYTON, S., CORPAS, M., RAJAN, D., VAN VOOREN, S., MOREAU, Y., PETTETT, R. M. & CARTER, N. P. 2009. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet, 84, 524-33.
FUKUYAMA, M., OHNO, S., MAKIYAMA, T. & HORIE, M. 2015. Novel SCN10A variants associated with Brugada syndrome. Europace.
GARCIA-MOLINA, E., LACUNZA, J., RUIZ-ESPEJO, F., SABATER, M., GARCIA-ALBEROLA, A., GIMENO, J. R., CANIZARES, F., GARCIA, A., MARTINEZ, P., VALDES, M. & TOVAR, I. 2013. A study of the SCN5A gene in a cohort of 76 patients with Brugada syndrome. Clin Genet, 83, 530-8.
GOURRAUD, J. B., BARC, J., THOLLET, A., LE SCOUARNEC, S., LE MAREC, H., SCHOTT, J. J., REDON, R. & PROBST, V. 2016. The Brugada Syndrome: A Rare Arrhythmia Disorder with Complex Inheritance. Front Cardiovasc Med, 3, 9.
GU, W., ZHANG, F. & LUPSKI, J. R. 2008. Mechanisms for human genomic rearrangements. Pathogenetics, 1, 4.
HU, D., BARAJAS-MARTINEZ, H., PFEIFFER, R., DEZI, F., PFEIFFER, J., BUCH, T., BETZENHAUSER, M. J., BELARDINELLI, L., KAHLIG, K. M., RAJAMANI, S., DEANTONIO, H. J., MYERBURG, R. J., ITO, H., DESHMUKH, P., MARIEB, M., NAM, G. B., BHATIA, A., HASDEMIR, C., HAISSAGUERRE, M., VELTMANN, C., SCHIMPF, R., BORGGREFE, M., VISKIN, S. & ANTZELEVITCH, C. 2014. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol, 64, 66-79.
HUANG, N., LEE, I., MARCOTTE, E. M. & HURLES, M. E. 2010. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet, 6, e1001154.
JEEVARATNAM, K., GUZADHUR, L., GOH, Y. M., GRACE, A. A. & HUANG, C. L. 2016. Sodium channel haploinsufficiency and structural change in ventricular arrhythmogenesis. Acta Physiol (Oxf), 216, 186-202.
KAPPLINGER, J. D., TESTER, D. J., ALDERS, M., BENITO, B., BERTHET, M., BRUGADA, J., BRUGADA, P., FRESSART, V., GUERCHICOFF, A., HARRIS-KERR, C., KAMAKURA, S., KYNDT, F., KOOPMANN, T. T., MIYAMOTO, Y., PFEIFFER, R., POLLEVICK, G. D., PROBST, V., ZUMHAGEN, S., VATTA, M., TOWBIN, J. A., SHIMIZU, W., SCHULZE-BAHR, E., ANTZELEVITCH, C., SALISBURY, B. A., GUICHENEY, P., WILDE, A. A., BRUGADA, R., SCHOTT, J. J. & ACKERMAN, M. J. 2010. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm, 7, 33-46.
KENT, W. J. 2002. BLAT–the BLAST-like alignment tool. Genome Res, 12, 656-64.
KENT, W. J., SUGNET, C. W., FUREY, T. S., ROSKIN, K. M., PRINGLE, T. H., ZAHLER, A. M. & HAUSSLER, D. 2002. The human genome browser at UCSC. Genome Res, 12, 996-1006.
KOOPMANN, T. T., BEEKMAN, L., ALDERS, M., MEREGALLI, P. G., MANNENS, M. M., MOORMAN, A. F., WILDE, A. A. & BEZZINA, C. R. 2007. Exclusion of multiple candidate genes and large genomic rearrangements in SCN5A in a Dutch Brugada syndrome cohort. Heart Rhythm, 4, 752-5.
LI, H. & DURBIN, R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754-60.
LI, H., HANDSAKER, B., WYSOKER, A., FENNELL, T., RUAN, J., HOMER, N., MARTH, G., ABECASIS, G., DURBIN, R. & GENOME PROJECT DATA PROCESSING, S. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078-9.
MEREGALLI, P. G., TAN, H. L., PROBST, V., KOOPMANN, T. T., TANCK, M. W., BHUIYAN, Z. A., SACHER, F., KYNDT, F., SCHOTT, J. J., ALBUISSON, J., MABO, P., BEZZINA, C. R., LE MAREC, H. & WILDE, A. A. 2009. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm, 6, 341-8.
PALLANTE, B. A., GIOVANNONE, S., FANG-YU, L., ZHANG, J., LIU, N., KANG, G., DUN, W., BOYDEN, P. A. & FISHMAN, G. I. 2010. Contactin-2 expression in the cardiac Purkinje fiber network. Circ Arrhythm Electrophysiol, 3, 186-94.
PFEUFER, A., SANNA, S., ARKING, D. E., MULLER, M., GATEVA, V., FUCHSBERGER, C., EHRET, G. B., ORRU, M., PATTARO, C., KOTTGEN, A., PERZ, S., USALA, G., BARBALIC, M., LI, M., PUTZ, B., SCUTERI, A., PRINEAS, R. J., SINNER, M. F., GIEGER, C., NAJJAR, S. S., KAO, W. H., MUHLEISEN, T. W., DEI, M., HAPPLE, C., MOHLENKAMP, S., CRISPONI, L., ERBEL, R., JOCKEL, K. H., NAITZA, S., STEINBECK, G., MARRONI, F., HICKS, A. A., LAKATTA, E., MULLER-MYHSOK, B., PRAMSTALLER, P. P., WICHMANN, H. E., SCHLESSINGER, D., BOERWINKLE, E., MEITINGER, T., UDA, M., CORESH, J., KAAB, S., ABECASIS, G. R. & CHAKRAVARTI, A. 2009. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet, 41, 407-14.
RUAN, Y., LIU, N. & PRIORI, S. G. 2009. Sodium channel mutations and arrhythmias. Nat Rev Cardiol, 6, 337-48.
SOTOODEHNIA, N., ISAACS, A., DE BAKKER, P. I., DORR, M., NEWTON-CHEH, C., NOLTE, I. M., VAN DER HARST, P., MULLER, M., EIJGELSHEIM, M., ALONSO, A., HICKS, A. A., PADMANABHAN, S., HAYWARD, C., SMITH, A. V., POLASEK, O., GIOVANNONE, S., FU, J., MAGNANI, J. W., MARCIANTE, K. D., PFEUFER, A., GHARIB, S. A., TEUMER, A., LI, M., BIS, J. C., RIVADENEIRA, F., ASPELUND, T., KOTTGEN, A., JOHNSON, T., RICE, K., SIE, M. P., WANG, Y. A., KLOPP, N., FUCHSBERGER, C., WILD, S. H., MATEO LEACH, I., ESTRADA, K., VOLKER, U., WRIGHT, A. F., ASSELBERGS, F. W., QU, J., CHAKRAVARTI, A., SINNER, M. F., KORS, J. A., PETERSMANN, A., HARRIS, T. B., SOLIMAN, E. Z., MUNROE, P. B., PSATY, B. M., OOSTRA, B. A., CUPPLES, L. A., PERZ, S., DE BOER, R. A., UITTERLINDEN, A. G., VOLZKE, H., SPECTOR, T. D., LIU, F. Y., BOERWINKLE, E., DOMINICZAK, A. F., ROTTER, J. I., VAN HERPEN, G., LEVY, D., WICHMANN, H. E., VAN GILST, W. H., WITTEMAN, J. C., KROEMER, H. K., KAO, W. H., HECKBERT, S. R., MEITINGER, T., HOFMAN, A., CAMPBELL, H., FOLSOM, A. R., VAN VELDHUISEN, D. J., SCHWIENBACHER, C., O’DONNELL, C. J., VOLPATO, C. B., CAULFIELD, M. J., CONNELL, J. M., LAUNER, L., LU, X., FRANKE, L., FEHRMANN, R. S., TE MEERMAN, G., GROEN, H. J., WEERSMA, R. K., VAN DEN BERG, L. H., WIJMENGA, C., OPHOFF, R. A., NAVIS, G., RUDAN, I., SNIEDER, H., WILSON, J. F., PRAMSTALLER, P. P., SISCOVICK, D. S., WANG, T. J., GUDNASON, V., VAN DUIJN, C. M., FELIX, S. B., FISHMAN, G. I., JAMSHIDI, Y., STRICKER, B. H., et al. 2010. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet, 42, 1068-76.
THORVALDSDOTTIR, H., ROBINSON, J. T. & MESIROV, J. P. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform, 14, 178-92.
TRUJILLO-QUINTERO, J. P., GUTIERREZ-AGULLO, M., OCHOA, J. P., MARTINEZ-MARTINEZ, J. G., DE UNA, D. & GARCIA-FERNANDEZ, A. 2018. Familial Brugada Syndrome Associated With a Complete Deletion of the SCN5A and SCN10A Genes. Rev Esp Cardiol (Engl Ed).
VAN DEN BOOGAARD, M., SMEMO, S., BURNICKA-TUREK, O., ARNOLDS, D. E., VAN DE WERKEN, H. J., KLOUS, P., MCKEAN, D., MUEHLSCHLEGEL, J. D., MOOSMANN, J., TOKA, O., YANG, X. H., KOOPMANN, T. T., ADRIAENS, M. E., BEZZINA, C. R., DE LAAT, W., SEIDMAN, C., SEIDMAN, J. G., CHRISTOFFELS, V. M., NOBREGA, M. A., BARNETT, P. & MOSKOWITZ, I. P. 2014. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest, 124, 1844-52.
VERKERK, A. O., REMME, C. A., SCHUMACHER, C. A., SCICLUNA, B. P., WOLSWINKEL, R., DE JONGE, B., BEZZINA, C. R. & VELDKAMP, M. W. 2012. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res, 111, 333-43.
WATSON, C. M., CRINNION, L. A., BERRY, I. R., HARRISON, S. M., LASCELLES, C., ANTANAVICIUTE, A., CHARLTON, R. S., DOBBIE, A., CARR, I. M. & BONTHRON, D. T. 2016. Enhanced diagnostic yield in Meckel-Gruber and Joubert syndrome through exome sequencing supplemented with split-read mapping. BMC Med Genet, 17, 1.
WECKSELBLATT, B. & RUDD, M. K. 2015. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet, 31, 587-99.
YANG, T., ATACK, T. C., STROUD, D. M., ZHANG, W., HALL, L. & RODEN, D. M. 2012. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res, 111, 322-32.
ZUMHAGEN, S., VELDKAMP, M. W., STALLMEYER, B., BAARTSCHEER, A., ECKARDT, L., PAUL, M., REMME, C. A., BHUIYAN, Z. A., BEZZINA, C. R. & SCHULZE-BAHR, E. 2013. A heterozygous deletion mutation in the cardiac sodium channel gene SCN5A with loss- and gain-of-function characteristics manifests as isolated conduction disease, without signs of Brugada or long QT syndrome. PLoS One, 8, e67963.